
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Цистична фиброза
Медицински стручњак за чланак

Последње прегледано: 04.07.2025
Цистична фиброза је генетска аутозомно рецесивна моногена болест коју карактерише поремећај секреције егзокриних жлезда виталних органа са оштећењем првенствено респираторног и дигестивног система, тешким током и неповољном прогнозом.
 [ 1 ]
[ 1 ]
Епидемиологија
Учесталост цистичне фиброзе варира између 1:2.500 и 1:4.600 новорођенчади. Сваке године се широм света роди око 45.000 људи са цистичном фиброзом. Учесталост носилаца гена цистичне фиброзе је 3-4%, при чему је око 275 милиона људи широм света носилаца овог гена, од чега око 5 милиона живи у Русији, а око 12,5 милиона у земљама ЗНД.
Узроци цистична фиброза
Цистична фиброза се преноси аутозомно рецесивно. Ген за цистичну фиброзу налази се у аутозому 7, садржи 27 ексона и састоји се од 250.000 парова нуклеотида.
Један ген може имати много мутација, од којих је свака специфична за одређену популацију или географски регион. Описано је више од 520 мутација, од којих је најчешћа делта-П-508, тј. супституција аминокиселине фенилаланина на позицији 508.
Патогенеза
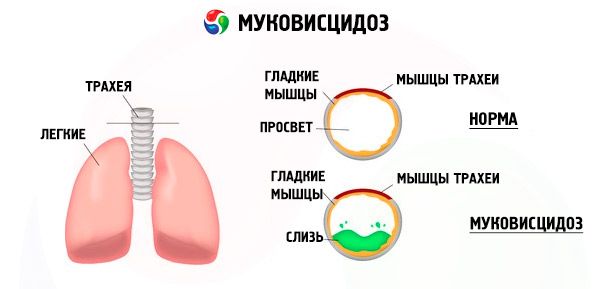
Мутације у гену цистичне фиброзе нарушавају структуру и функцију протеина који се зове CFTR (трансмембрански регулатор цистичне фиброзе). Овај протеин делује као хлоридни канал који учествује у размени воде и електролита епителних ћелија бронхопулмоналног система, гастроинтестиналног тракта, панкреаса, јетре и репродуктивног система. Као резултат поремећаја функције и структуре CFTR протеина, хлоридни јони Cl⁻ се акумулирају унутар ћелије. То доводи до промене електричног потенцијала у лумену изводних канала, што олакшава проток великих количина натријумових јона (Na⁺ ) из лумена канала у ћелију и додатно побољшава апсорпцију воде из перицелуларног простора.
Као резултат ових промена, тајна већине егзокриних жлезда се згушњава, њена евакуација је поремећена, што доводи до изражених секундарних поремећаја у органима и системима, најизраженијих у бронхопулмоналном и дигестивном систему.
У бронхијама се развија хронични инфламаторни процес различитог интензитета, функција цилијарног епитела је оштро поремећена, спутум постаје веома вискозан, густ, веома тешко се евакуише, примећује се његова стагнација, формирају се бронхиоло- и бронхиектазије, које временом постају све чешће. Ове промене доводе до повећања хипоксије и формирања хроничне плућне болести срца.
Пацијенти са цистичном фиброзом су изузетно предиспонирани за развој хроничне упале у бронхопулмоналном систему. То је због изражених поремећаја у локалном бронхопулмоналном одбрамбеном систему (смањени нивои IgA, интерферона, фагоцитне функције алвеоларних макрофага и леукоцита).
Алвеоларни макрофаги играју главну улогу у развоју хроничне упале у бронхопулмоналном систему. Они производе велике количине IL-8, што драматично повећава хемотаксију неутрофила у бронхијалном стаблу. Неутрофили се акумулирају у великим количинама у бронхијама и, заједно са епителним ћелијама, луче многе проинфламаторне цитокине, укључујући IL-1, 8, 6, фактор туморске некрозе и леукотриене.
Важну улогу у патогенези оштећења бронхопулмоналног система игра и висока активност ензима еластазе. Прави се разлика између егзогене и ендогене еластазе. Прву производи бактеријска флора (посебно Pseudomonas aeruginosa), другу - неутрофилни леукоцити. Еластаза уништава епител и друге структурне елементе бронхија, што доприноси даљем поремећају мукоцилијарног транспорта и брзом формирању бронхиектазије.
Неутрофилни леукоцити такође луче и друге протеолитичке ензиме. Алфа-1-антипирзин и секреторни инхибитор леукопротеиназа супротстављају се утицају протеолитичких ензима и стога штите бронхопулмонални систем од њиховог штетног утицаја. Међутим, нажалост, код пацијената са цистичном фиброзом, ови заштитни фактори су потиснути значајном количином неутрофилне протеазе.
Све ове околности доприносе уношењу инфекције у бронхопулмонални систем и развоју хроничног гнојног бронхитиса. Поред тога, треба узети у обзир да дефектни протеин који кодира ген цистичне фиброзе мења функционално стање бронхијалног епитела, што фаворизује адхезију бактерија на бронхијални епител, пре свега Pseudomonas aeruginosa.
Уз патологију бронхопулмоналног система, цистична фиброза такође узрокује тешка оштећења панкреаса, желуца, дебелог и танког црева, јетре.
Симптоми цистична фиброза
Цистична фиброза се манифестује различитим клиничким симптомима. Код новорођенчади, болест се може манифестовати меконијумским илеусом. Због недостатка или чак потпуног одсуства трипсина, меконијум постаје веома густ, вискозан и акумулира се у илеоцекалној регији. Даље се развија цревна опструкција, која се манифестује интензивним повраћањем са примесом жучи, надимањем стомака, недостатком излучивања меконијума, развојем симптома перитонитиса и брзим погоршањем клиничких манифестација синдрома тешке интоксикације. Дете може умрети у првим данима живота ако се не изврши хитна хируршка интервенција.
У мање тешким случајевима, карактеристичан знак цистичне фиброзе је обилна, честа столица, масна, са великом количином масти, са веома непријатним мирисом. Код 1/3 пацијената примећује се пролапс ректума.
Након тога, пацијенти настављају да доживљавају цревну дисфункцију, синдром малапсорпције, тешке поремећаје физичког развоја и тешку хиповитаминозу.
У првој или другој години живота јављају се симптоми оштећења бронхопулмоналног система (благи облик болести), што се манифестује кашљем који може бити изузетно изражен и подсећати на кашаљ код великог кашља. Кашаљ је праћен цијанозом, отежаним дисањем и одвајањем густог спутума, у почетку слузавог, а затим гнојног. Постепено се формира клиничка слика хроничног опструктивног бронхитиса и бронхиектазије, плућног емфизема и респираторне инсуфицијенције. Деца су изузетно подложна акутним респираторним вирусним и бактеријским инфекцијама, што доприноси егзацербацијама и прогресији бронхопулмоналне патологије. Могућ је развој бронхијалне астме зависне од инфекције.
Код деце школског узраста, цистична фиброза се може манифестовати као „цревне колике“. Пацијенти се жале на јаке пароксизмалне болове у стомаку, надимање и вишеструко повраћање. Приликом палпације стомака одређују се густе формације, које се налазе у пројекцији дебелог црева - фекалне масе помешане са густом, густом слузи. Деца су веома склона развоју хипохлоремијске алкалозе због прекомерног излучивања соли знојем по врућем времену, док се на кожи детета појављује „слани мраз“.
Болести бронхопулмоналног система код одраслих
Оштећење бронхопулмоналног система код пацијената са цистичном фиброзом (плућни облик болести) карактерише се развојем хроничног гнојног опструктивног бронхитиса, бронхиектазије, хроничне пнеумоније, плућног емфизема, респираторне инсуфицијенције и плућне болести срца. Неки пацијенти развијају пнеумоторакс и друге компликације цистичне фиброзе: ателектазу, плућне апсцесе, хемоптизу, плућно крварење и бронхијалну астму зависну од инфекције.
Пацијенти се жале на болан пароксизмални кашаљ са веома вискозним, тешко одвојивим мукопурулентним спутумом, понекад са примесом крви. Поред тога, изузетно је карактеристичан отежано дисање, прво током физичког напора, а затим и у мировању. Отежано дисање је узроковано бронхијалном опструкцијом. Многи пацијенти се жале на хронични ринитис изазван полипозом и синуситисом. Карактеристична је и значајна слабост, прогресивно смањење перформанси, честа акутна респираторна вирусна обољења. Приликом прегледа, скреће пажњу бледа кожа, отеченост лица, цијаноза видљивих слузокожа и јак отежано дисање. Са развојем декомпензоване плућне болести срца, појављује се едем у ногама. Може се приметити задебљање терминалних фаланги прстију у облику бубњака, а нокти у облику сатних стакала. Груди попримају облик бурета (због развоја плућног емфизема).
Перкусија плућа открива знаке емфизема - кутијасти звук, оштро ограничење покретљивости плућне ивице и спуштање доње ивице плућа. Аускултација плућа открива оштро дисање са продуженим издисајем, расуто суво хрипање и влажно средње и фино мехурасто хрипање. Код тешког емфизема плућа, дисање је оштро ослабљено.
Екстрапулмоналне манифестације цистичне фиброзе
Екстрапулмоналне манифестације цистичне фиброзе могу бити прилично изражене и јављају се често.
Оштећење панкреаса
Инсуфицијенција егзокрине функције панкреаса различитог степена тежине примећује се код 85% пацијената са цистичном фиброзом. Код мањег оштећења панкреаса, синдроми малдигестије и малапсорпције су одсутни, постоје само лабораторијске манифестације егзокрине инсуфицијенције (низак ниво трипсина и липазе у крви и садржају дванаестопалачног црева; често тешка стеатореја). Познато је да је за спречавање синдрома малдигестије довољно лучење само 1 до 2% укупне липазе. Клинички се манифестују само значајни поремећаји егзокрине функције.
У нормалним условима, ацинус панкреаса производе течни секрет богат ензимима. Како се секрет креће дуж изводног канала, обогаћује се водом и анјонима, те постаје још течнији. Код цистичне фиброзе, због поремећаја у структури и функцији трансмембранског регулатора (хлоридног канала), панкреатични секрет не добија довољну количину течности, постаје вискозан, а брзина његовог кретања дуж изводног канала се нагло успорава. Протеини секрета се таложе на зидовима малих изводних канала, што доводи до њихове блокаде. Како болест напредује, на крају се развија уништење и атрофија ацинуса - формира се хронични панкреатитис са егзокрином панкреатичном инсуфицијенцијом. То се клинички огледа у развоју синдрома малдигестије и малапсорпције. Панкреатична инсуфицијенција је главни узрок малапсорпције масти код цистичне фиброзе, али се то обично примећује код значајног недостатка липазе. Форшер и Дури (1991) указују да се у потпуном одсуству панкреасне липазе масти разграђују и апсорбују за 50-60%, што је последица присуства желудачних и пљувачних (сублингвалних) липаза, чија је активност близу доње границе норме. Уз поремећај разградње и апсорпције масти, долази и до поремећаја разградње и реапсорпције протеина. Око 50% протеина примљених храном губи се фецесом. Апсорпција угљених хидрата је погођена у мањој мери упркос недостатку α-амилазе, али метаболизам угљених хидрата може бити значајно поремећен.
Оштећење панкреаса се изражава у развоју синдрома малдигестије и малапсорпције са значајним губитком тежине и обилном масном столицом.
Развој синдрома малдигестије и малапсорпције такође је олакшан тешким поремећајем функције цревних жлезда, оштећеном секрецијом цревног сока и смањењем садржаја цревних ензима у њему.
Синдроми малдигестије и малапсорпције се називају и цревни облик цистичне фиброзе.
Оштећена ендокрина функција панкреаса (дијабетес мелитус) примећена је код пацијената са цистичном фиброзом у каснијим стадијумима болести (код 2% деце и 15% одраслих)
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Оштећење јетре и билијарних трактова
Код 13% пацијената са мешовитим и интестиналним облицима цистичне фиброзе развија се цироза јетре. Најтипичнија је за мутације W128X, делта-P508 и X1303K. Билијарна цироза јетре са порталном хипертензијом се открива код 5-10% пацијената. Према Велчу, Смиту (1995), клинички, морфолошки, лабораторијски, инструментални знаци оштећења јетре се откривају код 86% пацијената са цистичном фиброзом.
Многи пацијенти са цистичном фиброзом такође развијају хронични холециститис, често калкулозан.
Дисфункција полних жлезда
Пацијенти са цистичном фиброзом могу имати азооспермију, што је узрок неплодности. Смањена плодност је такође типична за жене.
Фазе
Постоје три степена тежине плућне цистичне фиброзе.
Благи облик цистичне фиброзе карактеришу ретке егзацербације (не више од једном годишње); током периода ремисије, клиничке манифестације су практично одсутне, а пацијенти су способни за рад.
Умерена тежина - егзацербације се примећују 2-3 пута годишње и трају око 2 месеца или дуже. У фази егзацербације јавља се интензиван кашаљ са тешко одвојивим спутумом, кратак дах чак и при мањем физичком напору, субфебрилна телесна температура, општа слабост, знојење. Истовремено, долази до кршења егзокрине функције панкреаса. У фази ремисије, радни капацитет није у потпуности обнављен, кратак дах током физичког напора остаје.
Тежак ток карактеришу веома честе егзацербације болести. Ремисије практично изостају. У клиничкој слици до изражаја долазе тешка респираторна инсуфицијенција, симптоми хроничне плућне болести срца, често декомпензоване, типична је хемоптиза. Примећује се значајан губитак тежине, пацијенти су потпуно инвалиди. По правилу, тешку бронхопулмоналну патологију прати оштро изражена дисфункција панкреаса.
Обрасци
- Бронхопулмоналне лезије
- Поновљена и рекурентна пнеумонија са продуженим током.
- Апсцесирајућа пнеумонија, посебно код одојчади.
- Хронична пнеумонија, посебно билатерална.
- Бронхијална астма отпорна на традиционалну терапију.
- Рекурентни бронхитис, бронхиолитис, посебно са културом Pseudomonas aeruginosa.
- Промене у гастроинтестиналном тракту
- Меконијумски илеус и његови еквиваленти.
- Синдром поремећене цревне апсорпције непознате генезе.
- Опструктивна жутица код новорођенчади са продуженим током.
- Цироза јетре.
- Дијабетес мелитус.
- Гастроезофагеални рефлукс.
- Холелитијаза.
- Ректални пролапс.
- Промене у другим органима и системима
- Поремећаји раста и развоја.
- Одложени сексуални развој.
- Мушка неплодност.
- Носни полипи.
- Браћа и сестре из породица са цистичном фиброзом.
[ 24 ]
Компликације и посљедице
Компликације из гастроинтестиналног тракта укључују:
- Дијабетес мелитус се развија код 8-12% пацијената старијих од 25 година.
- Фиброзирајућа колонопатија.
- Меконијумски илеус у неонаталном периоду (код 12% новорођенчади са цистичном фиброзом), синдром дисталне цревне опструкције, ректални пролапс, пептични улкус и гастроезофагеална рефлуксна болест.
Компликације јетре:
- Масна болест јетре (код 30-60% пацијената),
- Фокална билијарна цироза, мултинодуларна билијарна цироза и придружена портална хипертензија.
Портална хипертензија понекад доводи до смрти због варици једњака.
Преваленција холециститиса и жучних каменаца је већа код пацијената са цистичном фиброзом него код других особа.
Одложени пубертет и смањена плодност и друге компликације. Већина мушкараца има азооспермију и неразвијеност вас деференса.
Дијагностика цистична фиброза
Општа анализа крви - типична је анемија различитог степена тежине, обично нормо- или хипохромна. Анемија има полифакторијалну генезу (смањена апсорпција гвожђа и витамина Б12 у цревима због развоја синдрома малапсорпције). Могућа је леукопенија, са развојем гнојног бронхитиса и пнеумоније - леукоцитоза, повећана седиментација еритроцита.
Општа анализа урина - нема значајних промена, у ретким случајевима се примећује блага протеинурија.
Копрололошки преглед - примећују се стеатореја, креатореја. Бекер (1987) препоручује мерење химотрипсина и масних киселина у фецесу. Пре одређивања химотрипсина у фецесу, потребно је прекинути узимање дигестивних ензима најмање 3 дана пре прегледа. Код цистичне фиброзе, количина химотрипсина у фецесу је смањена, а количина масних киселина повећана (нормално излучивање масних киселина је мање од 20 ммол/дан). Потребно је узети у обзир да се повећано излучивање масних киселина фецесом примећује и код:
- недостатак коњугованих масних киселина у танком цреву због отказивања јетре, опструкције жучних канала, значајне бактеријске колонизације танког црева (у овом случају долази до интензивне хидролизе жучних киселина);
- илеитис;
- целијакија (са развојем синдрома малапсорпције);
- ентеритис;
- цревни лимфоми;
- Виплова болест;
- алергије на храну;
- убрзани транзит прехрамбених маса код дијареје различитог порекла, карциноидног синдрома, тиреотоксикозе.
Биохемијски тест крви - смањен ниво укупних протеина и албумина, повећан ниво алфа2 и гама глобулина, билирубина и аминотрансфераза (у случају оштећења јетре), смањена активност амилазе, липазе, трипсина и ниво гвожђа и калцијума (у случају развоја синдрома малдигестије, малапсорпције).
Анализа спутума - присуство великог броја неутрофилних леукоцита и микроорганизама (током бактериоскопије спутума).
Проучавање апсорпционе функције танког црева и егзокрине функције панкреаса открива значајне поремећаје.
Рентгенски преглед плућа - открива промене, чија тежина зависи од тежине и фазе болести. Најкарактеристичније промене су:
- повећано плућно обликовање услед перибронхијалних интерстицијалних промена;
- ширење корена плућа;
- слика лобуларне, субсегменталне или чак сегментне ателектазе плућа;
- повећана транспарентност плућних поља, углавном у горњим деловима, низак положај и недовољна покретљивост дијафрагме, ширење ретростерналног простора (манифестација плућног емфизема);
- сегментна или полисегментална инфилтрација плућног ткива (у развоју пнеумоније).
Бронхографија открива промене изазване опструкцијом бронхија вискозним спутумом (фрагментација пуњења бронхија контрастом, неравне контуре, феномен руптуре бронха, значајно смањење броја бочних грана), као и бронхоектазије (цилиндрични, мешовити), локализоване углавном у доњим деловима плућа.
Бронхоскопија открива дифузни гнојни бронхитис са обилним густим, вискозним спутумом и фибринозним филмовима.
Спирометрија - већ у раним фазама болести открива респираторну инсуфицијенцију опструктивног типа (смањење ФВК, ФЕВ1, Тифноов индекс), рестриктивну (смањење ФВК) или, најчешће, опструктивно-рестриктивну (смањење ФВК, ФВК, ФЕВ1, Тифноов индекс).
Гибсонов и Куков тест зноја (тест електролита у зноју) подразумева стимулисање знојења помоћу електрофорезе пилокарпина са накнадним одређивањем хлорида у зноју. Доерехук (1987) описује тест на следећи начин. Електрофореза пилокарпина се изводи на подлактици, електрична струја је 3 mA. Након чишћења коже дестилованом водом, зној се сакупља помоћу филтер папира који се поставља на стимулисано подручје, прекривен газом да би се спречило испаравање са њега. Након 30-60 минута, филтер папир се уклања и елуира у дестилованој води. Мери се количина сакупљеног зноја. Да би се добили поуздани резултати, потребно је сакупити најмање 50 мг (пожељно 100 мг) зноја.
Ако је концентрација хлорида већа од 60 ммол/л, дијагноза цистичне фиброзе се сматра вероватном; ако је концентрација хлорида већа од 100 ммол/л, она је поуздана; у овом случају, разлика у концентрацији хлора и натријума не би требало да пређе 8-10 ммол/л. Хадсон (1983) препоручује да се, ако је садржај натријума и хлорида у зноју граничан, изврши преднизолонски тест (5 мг орално током 2 дана, након чега следи одређивање електролита у зноју). Код особа које не пате од цистичне фиброзе, ниво натријума у зноју се смањује на доњу границу нормале; код цистичне фиброзе се не мења. Тест зноја се препоручује сваком детету са хроничним кашљем.
Анализа крвних мрља или узорака ДНК на главне мутације гена цистичне фиброзе је најосетљивији и најспецифичнији дијагностички тест. Међутим, ова метода је погодна само за земље у којима је стопа мутације делта-P508 већа од 80%. Поред тога, техника је веома скупа и технички сложена.
Пренатална дијагноза цистичне фиброзе се врши одређивањем изоензима алкалне фосфатазе у амнионској течности. Ова метода постаје могућа од 18-20 недеља трудноће.
Главни критеријуми за дијагнозу цистичне фиброзе су следећи:
- индикације у анамнези одложеног физичког развоја у детињству, рекурентних хроничних респираторних болести, диспептичних поремећаја и дијареје, присуства цистичне фиброзе код блиских рођака;
- хронични опструктивни бронхитис, често рекурентни, са развојем бронхиектазије и плућног емфизема, често рекурентна пнеумонија;
- хронични рекурентни панкреатитис са израженим смањењем егзокрине функције, синдром малапсорпције;
- повећан садржај хлора у зноју пацијента;
- неплодност са очуваном сексуалном функцијом.
Успешна дијагноза и диференцијална дијагноза цистичне фиброзе олакшава се идентификацијом ризичних група.
Програм скрининга за цистичну фиброзу
- Општа анализа крви, урина, спутума.
- Бактериолошка анализа спутума.
- Копролошка анализа.
- Биохемијски тест крви: одређивање укупних протеина и протеинских фракција, глукозе, билирубина, аминотрансфераза, алкалне фосфатазе, гама-глутамил транспептидазе, калијума, калцијума, гвожђа, липазе, амилазе, трипсина.
- Проучавање егзокрине функције панкреаса и апсорпционе функције црева.
- Флуороскопија и радиографија плућа, ЦТ скенирање плућа.
- ЕКГ.
- Ехокардиографија.
- Бронхоскопија и бронхографија.
- Спирометрија.
- Тест знојења.
- Консултације са генетичарем.
- Анализа крвних мрља или узорака ДНК за главне мутације гена цистичне фиброзе.
Како испитивати?
Који су тестови потребни?
Кога треба контактирати?
Третман цистична фиброза
Врста и тежина симптома цистичне фиброзе могу значајно да варирају, тако да не постоји типичан план лечења; он је индивидуализован за сваку особу.
Терапија се састоји од следећих терапеутских мера:
- Вежбе дисања и постурална дренажа помажу у уклањању густог слузи који се накупља у плућима. Неке технике чишћења дисајних путева захтевају помоћ чланова породице, пријатеља или пулмолога. Многи људи користе надувни прслук за груди који вибрира на високој фреквенцији.

- Инхалациони лекови који имају бронходилататорне, дренажне (муколитици) и антибактеријске ефекте (на пример, флуорокинолони).
- Препарати који садрже панкреасне ензиме за побољшање варења. Ови препарати се узимају током оброка.
- Мултивитамини (укључујући витамине растворљиве у мастима).
Године 2015, FDA је одобрила други лек за лечење цистичне фиброзе који циља дефектни протеин познат као CFTR. Први лек, такозвани CFTR модулатор, одобрен је 2012. године. Очекује се да ће CFTR модулатори продужити живот неких људи са цистичном фиброзом за деценије.
За лечење следећих респираторних компликација може бити потребна операција:
- Пнеумоторакс, масивна рекурентна или перзистентна хемоптиза, назални полипи, перзистентни и хронични синуситис.
- Меконијумски илеус, инвагинација, ректални пролапс.
Трансплантација плућа се врши у терминалној фази болести.
Прогноза
Просечна старост преживљавања пацијената са цистичном фиброзом креће се од 35 до 40 година. Просечна старост преживљавања је виша код мушкараца него код жена.
Са савременим стратегијама лечења, 80% пацијената доживи одрасло доба. Међутим, цистична фиброза значајно ограничава функционалне способности пацијента. Још увек не постоји лек за ову болест.