
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Шарко-Мари-Тут болест.
Медицински стручњак за чланак

Последње прегледано: 12.07.2025
Перонеална мишићна атрофија, Шарко-Мари-Тутов синдром или болест је група хроничних наследних болести са оштећењем периферних нерава.
Према МКБ-10, у одељку о болестима нервног система, шифра за ову болест је Г60.0 (наследна моторна и сензорна неуропатија). Такође је укључена у листу болести које се не лече као ричади.
Епидемиологија
Према клиничкој статистици, преваленција свих врста болести Шарко-Мари-Тута на 100 хиљада становника је 19 случајева (према другим изворима, један случај на 2,5-10 хиљада становника).
ЦМТ тип 1 чини око две трећине случајева (један случај на 5.000 до 7.000 становника), а скоро 70% њих је повезано са дуплирањем гена ПМП22. Више од 1,2 милиона људи широм света пати од ове врсте болести.
Учесталост карцинома меланоме типа 4 процењује се на 1-5 случајева на 10.000 деце. [ 1 ]
Узроци Шарко-Мари-Тут болест
Према класификацији полинеуропатских синдрома, перонеална (фибуларна) мишићна атрофија, Шарко-Мари-Тутова неурална амиотрофија или Шарко-Мари-Тутова болест (скраћено CMT) односи се на генетски одређене моторно-сензорне полинеуропатије. [ 2 ]
То јест, узроци његовог настанка су генетске мутације. И у зависности од природе генетских одступања, разликују се главни типови или врсте овог синдрома: демијелинизирајући и аксонални. Прва група обухвата Шарко-Мари-Тутову болест типа 1 (CMT1), која настаје због дупликације гена PMP22 на хромозому 17, који кодира трансмембрански периферни мијелински протеин 22. Као резултат тога, долази до сегментне демијелинације аксонског омотача (процеса нервних ћелија) и смањења брзине проводљивости нервног сигнала. Поред тога, мутације могу бити у неким другим генима.
Аксонални облик је Шарко-Мари-Тутова болест типа 2 (CMT2), која погађа саме аксоне и повезана је са патолошким променама у гену MFN2 на локусу 1p36.22, који кодира мембрански протеин митофузин-2, који је неопходан за фузију митохондрија и формирање функционалних митохондријалних мрежа унутар периферних нервних ћелија. Постоји више од десетак подтипова CMT2 (са мутацијама у специфичним генима).
Треба напоменути да је тренутно идентификовано више од стотину гена чија оштећења, пренета наслеђивањем, узрокују различите подтипове Шарко-Мари-Тутове болести. На пример, мутације у гену RAB7 доводе до ЦМТ типа 2Б; промена гена SH3TC2 (који кодира један од мембранских протеина Шванових ћелија) узрокује ЦМТ тип 4Ц, који се манифестује у детињству и карактерише се демијелинацијом моторних и сензорних неурона (постоји десетак и по облика типа 4 ове болести).
Ретки карцином меланоме типа 3 (назван Дежерин-Сотасов синдром) почиње да се развија у раном детињству и узрокован је мутацијама у генима PMP22, MPZ, EGR2 и другим.
Када се ЦМТ тип 5 појави у доби од 5-12 година, не примећује се само моторна неуропатија (у облику спастичне парапарезе доњих екстремитета), већ и оштећење оптичких и слушних живаца.
Мишићна слабост и оптичка атрофија (са губитком вида), као и проблеми са равнотежом, типични су за ЦМТ тип 6. А код Шарко-Мари-Тутове болести типа 7 не постоји само моторно-сензорна неуропатија, већ и болест мрежњаче у облику пигментозе ретинитиса.
Чешћа међу мушкарцима, X-везана каротидна мелитусна болест или Шарко-Мари-Тут болест са тетрапарезом удова (слабост покрета и руку и ногу) је демијелинизирајућег типа и сматра се да је резултат мутације у гену GJB1 на дугом краку X хромозома, који кодира конексин 32, трансмембрански протеин Шванових ћелија и олигодендроцита који регулише пренос нервних сигнала. [ 3 ]
Фактори ризика
Главни фактор ризика за ЦМТ је породична историја болести, односно код блиских рођака.
Према генетичарима, ако су оба родитеља носиоци аутозомно рецесивног гена за Шарко-Мари-Тутову болест, ризик од рађања детета које ће развити ову болест је 25%. А ризик да ће дете бити носилац овог гена (али неће имати никакве симптоме) процењује се на 50%.
У случају X-везаног наслеђивања (када се мутирани ген налази на X хромозому жене), постоји 50% ризика да ће мајка пренети ген на свог сина, који ће развити карциномску меланоцитну склерозу (CMT). Болест се можда неће јавити када се роди женско дете, али ћеркини синови (унуци) могу наследити дефектни ген, а болест ће се развити.
Патогенеза
Код било које врсте Шарко-Мари-Тутовог синдрома, његова патогенеза је узрокована наследном аномалијом периферних нерава: моторних (покретних) и сензорних (сензитивних).
Ако је ЦМТ тип демијелинизирајући, онда уништење или дефект мијелинског омотача који штити аксоне периферних нерава доводи до успоравања преноса нервних импулса у периферном нервном систему - између мозга, мишића и чулних органа.
Код аксоналног типа болести, сами аксони су погођени, што негативно утиче на јачину нервних сигнала, која је недовољна за потпуну стимулацију мишића и сензорних органа.
Такође прочитајте:
Како се преноси Шарко-Мари-Тут синдром? Дефектни гени могу се наслеђивати аутозомно доминантно или аутозомно рецесивно.
Најчешћи тип наслеђивања, аутозомно доминантно, јавља се када постоји једна копија мутираног гена (који носи један од родитеља). А вероватноћа преношења карцинома меланоме (CMT) на свако рођено дете процењује се на 50%. [ 4 ]
Код аутозомно рецесивног наслеђивања, потребне су две копије дефектног гена (по једна од сваког родитеља који не показује никакве знаке болести) да би се болест развила.
У 40-50% случајева јавља се аутозомно доминантна наследна демијелинација, тј. карцинома меланоцитне болести (CMT) тип 1; у 12-26% случајева, аксонална CMT, тј. тип 2. А у 10-15% случајева примећује се X-везано наслеђивање. [ 5 ]
Симптоми Шарко-Мари-Тут болест
Типично, први знаци ове болести почињу да се јављају у детињству и адолесценцији и постепено се развијају током живота, мада се синдром може појавити касније. Комбинација симптома је променљива, а брзина прогресије болести, као и њена тежина, немогуће је предвидети.
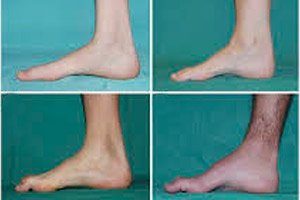
Типични симптоми почетне фазе укључују повећан општи умор; смањен тонус (слабост) мишића стопала, скочних зглобова и потколеница; недостатак рефлекса. Ово отежава кретање стопала и доводи до дисбазије (поремећаја хода) у виду вишег подизања ногу, често са честим спотицањима и падовима. Знаци Шарко-Мари-Тутове болести код малог детета могу укључивати изражену неспретност и тешкоће у ходању некарактеристичне за узраст, повезане са билатералним спуштањем стопала. Деформације стопала су такође карактеристичне: висок свод (шупље стопало) или тешка равна стопала, закривљени (чекићасти) прсти.
У случају ходања на прстима на позадини мишићне хипотоније, неуролог може посумњати да дете има ЦМТ типа 4, код којег деца можда неће моћи да ходају до адолесценције.
Како болест напредује, атрофија мишића и слабост се шире на горње екстремитете, што отежава извођење фине моторике и обављање нормалних задатака рукама. Смањење тактилних сензација и способност осећаја топлоте и хладноће, као и утрнулост у стопалима и рукама, указују на оштећење аксона сензорних нерава.
Код Шарко-Мари-Тутове болести типа 3 и 6, која се манифестује у детињству, примећују се сензорна атаксија (поремећај координације покрета и равнотеже), трзање мишића и тремор, оштећење фацијалног нерва, атрофија оптичког нерва са нистагмусом и губитак слуха.
У каснијим фазама може доћи до неконтролисаног тремора (тремора) и честих грчева мишића; проблеми са кретањем могу довести до развоја бола: мишићног, зглобног, неуропатског.
Компликације и посљедице
Шарко-Мари-Тут болест може имати компликације и последице као што су:
- чешћа уганућа и преломи;
- контрактуре повезане са скраћивањем периартикуларних мишића и тетива;
- сколиоза (закривљеност кичме);
- проблеми са дисањем – када су оштећена нервна влакна која инервишу мишиће дијафрагме:
- губитак способности самосталног кретања.
Дијагностика Шарко-Мари-Тут болест
Дијагноза обухвата клинички преглед, анамнезу (укључујући породичну историју), неуролошки и системски преглед.
Тестови се изводе ради провере обима покрета, осетљивости и тетивних рефлекса. Нервна проводљивост може се проценити инструменталном дијагностиком – електромиографијом или електронеуромиографијом. Такође могу бити потребни ултразвук или магнетна резонанца. [ 6 ]
Генетско или ДНК тестирање за откривање најчешћих генетских мутација које узрокују карциномску меланоцитну мутацију (CMT) у узорку крви је ограничено јер ДНК тестови тренутно нису доступни за све врсте болести. За више информација погледајте Генетско тестирање.
У неким случајевима се врши биопсија периферног нерва (обично суралног нерва).
Диференцијална дијагноза
Диференцијална дијагноза укључује друге периферне неуропатије, Душенову мишићну дистрофију, мијелопатске и мијастеничне синдроме, дијабетичку неуропатију, мијелопатије код мултипле и амиотрофичне латералне склерозе, Гилен-Бареов синдром, трауму перонеалног нерва и његову атрофију (укључујући и стезање између лумбалних дискова кичме), оштећење малог мозга или таламуса, као и нежељене ефекте хемотерапије (током лечења цитостатицима као што су винкристин или паклитаксел). [ 7 ]
Кога треба контактирати?
Третман Шарко-Мари-Тут болест
Данас, лечење ове наследне болести укључује терапију вежбања (усмерену на јачање и истезање мишића); радну терапију (која помаже пацијентима са слабошћу мишића у рукама); и употребу ортопедских помагала за олакшавање ходања. Ако је потребно, прописују се лекови против болова или антиконвулзиви. [ 8 ]
У случајевима тешких равних стопала може се извршити остеотомија, а у случају деформације пете индицирана је хируршка корекција – артродеза. [ 9 ]
У току су истраживања како генетске компоненте болести, тако и метода њеног лечења. Употреба матичних ћелија, одређених хормона, лецитина или аскорбинске киселине још увек није дала позитивне резултате.
Али захваљујући недавним истраживањима, нешто ново се заиста може појавити у лечењу Шарко-Мари-Тутове болести у блиској будућности. Тако је од 2014. године француска компанија Pharnext развијала, а од средине 2019. године су у току клиничка испитивања лека PXT3003 за лечење ЦМТ типа 1 код одраслих, који сузбија повећану експресију гена PMP22, побољшава мијелинацију периферних нерава и ублажава неуромускуларне симптоме.
Специјалисти медицинске компаније Sarepta Therapeutics (САД) раде на стварању генске терапије за Шарко-Мари-Тутову болест типа 1. Ова терапија ће користити безопасни адено-асоцирани вирус (AAV) из рода Dependovirus са линеарним једноланчаним ДНК геномом, који ће пренети ген NTF3 у тело, кодирајући протеин неуротрофин-3 (NT-3) неопходан за функционисање Шванових нервних ћелија.
Хеликсмит ће до краја 2020. године започети клиничка испитивања генске терапије Енгенсис (VM202), коју је развила Јужна Кореја, за лечење мишићних симптома код каротидне мишићне мачака типа 1. [ 10 ]
Превенција
Превенција ХМТ може бити генетско саветовање будућих родитеља, посебно ако неко у пару има породичну историју болести. Међутим, идентификовани су случајеви de novo тачкастих генских мутација, односно у одсуству болести у породичној историји.
Током трудноће, вероватноћа болести Шарко-Мари-Тута код будућег детета може се проверити биопсијом хорионских ресица (од 10. до 13. недеље гестације), као и анализом амнионске течности (у 15-18. недељи).
Прогноза
Генерално, прогноза за различите врсте Шарко-Мари-Тутовог обољења зависи од клиничке тежине, али у свим случајевима болест напредује споро. Многи пацијенти имају инвалидитет, иако то не смањује очекивани животни век.