
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Наследни нефритис (Алпортов синдром) код деце
Медицински стручњак за чланак

Последње прегледано: 05.07.2025
Наследни нефритис (Алпортов синдром) је генетски одређена наследна неимуна гломерулопатија, која се манифестује хематуријом (понекад са протеинуријом), прогресивним падом бубрежне функције са развојем хроничне бубрежне инсуфицијенције, често комбинованом са сензоринеуралном глувоћом и оштећењем вида.
Болест је први пут описана 1902. године од стране Л. Г. Гатрија, који је посматрао породицу у којој је хематурија примећена у неколико генерација. Године 1915, А. Ф. Херст је описао развој уремије код чланова исте породице. Године 1927, А. Алпорт је први идентификовао губитак слуха код неколико рођака са хематуријом. Педесетих година 20. века описане су очне лезије код сличне болести. Године 1972, код пацијената са наследном хематуријом, током морфолошке студије бубрежног ткива, Хинглаис и сарадници су открили неравномерно ширење и стратификацију гломеруларних базалних мембрана. Године 1985, идентификована је генетска основа наследног нефритиса - мутација у гену за колаген типа IV (Fiengold et al., 1985).
Проучавање генетске природе болести омогућило нам је да закључимо да су разлике у фенотипским манифестацијама наследног нефритиса (са или без губитка слуха) последица степена експресије мутантног гена. Стога се тренутно све клиничке варијанте сматрају манифестацијама једне болести, а термин „наследни нефритис“ је синоним за термин „Алпортов синдром“.
Према епидемиолошким студијама, наследни нефритис се јавља са учесталошћу од 17 на 100.000 деце.
 [
[ Узроци Алпортовог синдрома
Генетска основа болести је мутација у гену а-5 ланца колагена типа IV. Овај тип је универзалан за базалне мембране бубрега, кохлеарни апарат, капсулу сочива, мрежњачу и рожњачу ока, што је доказано у студијама које користе моноклонска антитела против ове фракције колагена. Недавно је назначена могућност коришћења ДНК сонди за пренаталну дијагностику наследног нефритиса.
Истиче се значај тестирања свих чланова породице ДНК сондама ради идентификације носилаца мутантног гена, што је од великог значаја у спровођењу медицинског и генетског саветовања породица са овом болешћу. Међутим, до 20% породица нема рођаке који пате од болести бубрега, што указује на високу учесталост спонтаних мутација абнормалног гена. Већина пацијената са наследним нефритисом има у својим породицама особе са болестима бубрега, губитком слуха и патологијом вида; важни су крвни бракови између људи са једним или више предака, јер се у браку сродних особа повећава вероватноћа пријема истих гена од оба родитеља. Утврђени су аутозомно доминантни, аутозомно рецесивни и доминантни, X-везани путеви преноса.
Код деце се најчешће разликују три типа наследног нефритиса: Алпортов синдром, наследни нефритис без губитка слуха и фамилијарна бенигна хематурија.
Алпортов синдром је наследни нефритис са оштећењем слуха. Заснован је на комбинованом дефекту у структури колагена гломеруларне базалне мембране бубрега, структура уха и ока. Ген класичног Алпортовог синдрома налази се у локусу 21-22 q дугог крака X хромозома. У већини случајева наслеђује се на доминантан начин, повезан са X хромозомом. У том смислу, Алпортов синдром је тежи код мушкараца, јер је код жена функција мутантног гена компензована здравим алелом другог, неоштећеног хромозома.
Генетска основа за развој наследног нефритиса су мутације у генима алфа ланаца колагена типа IV. Познато је шест алфа ланаца колагена типа IV G: гени а5- и а6-ланаца (Col4A5 и Col4A5) налазе се на дугом краку X хромозома у зони 21-22q; гени а3- и а4-ланаца (Col4A3 и Col4A4) налазе се на 2. хромозому; гени а1- и а2-ланаца (Col4A1 и Col4A2) налазе се на 13. хромозому.
У већини случајева (80-85%), детектује се X-везани образац наслеђивања болести, повезан са оштећењем гена Col4A5 као резултат делеције, тачкастих мутација или поремећаја сплајсинга. Тренутно је пронађено више од 200 мутација гена Col4A5, одговорних за поремећај синтезе a5-ланаца колагена типа IV. Код овог типа наслеђивања, болест се манифестује код деце оба пола, али код дечака је тежа.
Мутације у локусима гена Col4A3 и Col4A4 одговорних за синтезу а3 и а4 ланаца колагена типа IV наслеђују се аутозомно. Према истраживањима, аутозомно доминантни тип наслеђивања примећен је код 16% случајева наследног нефритиса, а аутозомно рецесивни тип код 6% пацијената. Познато је око 10 варијанти мутација гена Col4A3 и Col4A4.
Резултат мутација је кршење процеса склапања колагена типа IV, што доводи до кршења његове структуре. Колаген типа IV је једна од главних компоненти гломеруларне базалне мембране, кохлеарног апарата и сочива ока, чија ће патологија бити откривена у клиници наследног нефритиса.
Колаген типа IV, који је део гломеруларне базалне мембране, састоји се углавном од два a1-ланца (IV) и једног a2-ланца (IV), а садржи и a3, a4, a5-ланце. Најчешће, код X-везаног наслеђивања, мутација гена Col4A5 је праћена одсуством a3-, a4-, a5- и a6-ланаца у структури колагена типа IV, а број o1- и a2-ланаца у гломеруларној базалној мембрани се повећава. Механизам ове појаве није јасан, претпоставља се да су узрок посттранскрипционе промене у mRNA.
Одсуство а3, а4 и а5 ланаца у структури колагена типа IV гломеруларних базалних мембрана доводи до њиховог истањивања и крхкости у раним фазама Алпортовог синдрома, што се клинички чешће манифестује хематуријом (ређе хематуријом са протеинуријом или само протеинуријом), губитком слуха и лентиконусом. Даља прогресија болести доводи до задебљања и оштећене пропустљивости базалних мембрана у каснијим фазама болести, са пролиферацијом колагена типа V и VI у њима, што се манифестује повећањем протеинурије и смањењем бубрежне функције.
Природа мутације која је у основи наследног нефритиса у великој мери одређује његову фенотипску манифестацију. У случају делеције X хромозома са истовременом мутацијом гена Col4A5 и Col4A6 одговорних за синтезу а5- и а6-ланаца колагена типа IV, Алпортов синдром је комбинован са леиомиоматозом једњака и гениталија. Према подацима истраживања, у случају мутације гена Col4A5 повезане са делецијом, примећује се већа тежина патолошког процеса, комбинација оштећења бубрега са екстрареналним манифестацијама и раним развојем хроничне бубрежне инсуфицијенције, у поређењу са тачкастом мутацијом овог гена.
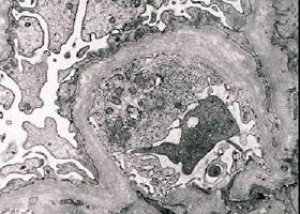
Морфолошки, електронска микроскопија открива истањивање и стратификацију гломеруларних базалних мембрана (посебно ламина денса) и присуство електронски густих гранула. Гломеруларне лезије могу бити хетерогене код истог пацијента, од минималних фокалних мезангијалних лезија до гломерулосклерозе. Гломерулитис код Алпортовог синдрома је увек имунонегативан, што га разликује од гломерулонефритиса. Карактеристични фактори укључују развој тубуларне атрофије, лимфохистиоцитну инфилтрацију и присуство „пенастих ћелија“ са липидним инклузијама – липофага. Како болест напредује, открива се задебљање и изражено уништавање гломеруларних базалних мембрана.
Откривене су одређене промене у имунолошком систему. Пацијенти са наследним нефритисом имају смањен ниво Ig А и тенденцију ка повећању концентрације IgM у крви, ниво IgG може бити повећан у раним фазама болести, а смањен у каснијим фазама. Можда је повећање концентрације IgM и G врста компензаторне реакције као одговор на недостатак IgA.
Функционална активност Т-лимфоцитног система је смањена; примећује се селективно смањење Б-лимфоцита одговорних за синтезу Ig А, фагоцитна веза имунитета је поремећена, углавном због поремећаја хемотаксије и интрацелуларних процеса варења у неутрофилима
Приликом испитивања биопсије бубрега код пацијената са Алпортовим синдромом, подаци електронске микроскопије откривају ултраструктурне промене у гломеруларној базалној мембрани: проређивање, поремећај структуре и цепање гломеруларних базалних мембрана са променом њене дебљине и неравним контурама. У раним фазама наследног нефритиса, дефект одређује проређивање и крхкост гломеруларних базалних мембрана.
Истанчавање гломеруларних мембрана је повољнији знак и чешћи је код девојчица. Константнији електронско-микроскопски знак код наследног нефритиса је цепање базалне мембране, а тежина њеног уништења корелира са тежином процеса.
Симптоми Алпортовог синдрома код деце
Први симптоми Алпортовог синдрома у облику изолованог уринарног синдрома најчешће се откривају код деце прве три године живота. У већини случајева, болест се открива случајно. Уринарни синдром се открива током превентивног прегледа детета, пре пријема у установу за бригу о деци или током АРВИ. У случају патологије у урину током АРВИ. Код наследног нефритиса, за разлику од стеченог гломерулонефритиса, нема латентног периода.
У почетној фази болести, здравље детета мало пати, карактеристична особина је перзистентност и отпорност уринарног синдрома. Један од главних знакова је хематурија различитог степена тежине, примећена у 100% случајева. Повећање степена хематурије се примећује током или након респираторних инфекција, физичке активности или након превентивних вакцинација. Протеинурија у већини случајева не прелази 1 г/дан, на почетку болести може бити непостојана, како процес напредује, протеинурија се повећава. Периодично, леукоцитурија са превлашћу лимфоцита може бити присутна у уринарном седименту, што је повезано са развојем интерстицијалних промена.
Накнадно, делимична бубрежна функција је оштећена, опште стање пацијента се погоршава: јављају се интоксикација, мишићна слабост, артеријска хипотензија, често се јавља оштећење слуха (посебно код дечака), а понекад и оштећење вида. Интоксикација се манифестује бледилом, умором и главобољама. У почетној фази болести, губитак слуха се у већини случајева открива само аудиографијом. Губитак слуха код Алпортовог синдрома може се јавити у различитим периодима детињства, али најчешће се губитак слуха дијагностикује у узрасту од 6-10 година. Губитак слуха код деце почиње високим фреквенцијама, достижући значајан степен у ваздушној и коштаној проводљивости, прелазећи из звучно-проводног у звучно-перцептивни губитак слуха. Губитак слуха може бити један од првих симптома болести и може претходити уринарном синдрому.
У 20% случајева, пацијенти са Алпортовим синдромом имају промене у органима вида. Најчешће откривене аномалије су оне сочива: сферофокија, предњи, задњи или мешовити лентиконус и разне катаракте. У породицама са Алпортовим синдромом постоји значајна учесталост миопије. Више истраживача стално примећује билатералне перимакуларне промене у овим породицама у облику светлих беличастих или жућкастих гранулација у жутом телу. Они сматрају да је овај знак стални симптом који има високу дијагностичку вредност код Алпортовог синдрома. К. С. Чуг и др. (1993) у офталмолошкој студији пронашли су код пацијената са Алпортовим синдромом смањење оштрине вида у 66,7% случајева, предњи лентиконус у 37,8%, ретиналне пеге у 22,2%, катаракту у 20% и кератоконус у 6,7%.
Код неке деце са наследним нефритисом, посебно када се развије бубрежна инсуфицијенција, примећује се значајно заостајање у физичком развоју. Како бубрежна инсуфицијенција напредује, развија се артеријска хипертензија. Код деце се чешће открива у адолесценцији и у старијим старосним групама.
Пацијенти са наследним нефритисом карактеришу се присуством различитих (више од 5-7) стигми везивног ткива дисморфогенезе. Међу стигмама везивног ткива код пацијената најчешћи су хипертелоризам очију, високо непце, аномалије загриза, абнормални облик ушних шкољки, закривљеност малог прста на рукама и „сандални размак“ на стопалима. Наследни нефритис карактерише униформност стигми дисморфогенезе унутар породице, као и висока учесталост њихове дистрибуције међу рођацима пробанда по чијој се линији болест преноси.
У раним стадијумима болести детектује се изоловано смањење парцијалних бубрежних функција: транспорт аминокиселина, електролита, концентрациона функција, ацидогенеза, касније промене утичу на функционално стање и проксималног и дисталног дела нефрона и карактеришу се комбинованим парцијалним поремећајима. Смањење гломеруларне филтрације јавља се касније, чешће у адолесценцији. Како наследни нефритис напредује, развија се анемија.
Дакле, наследни нефритис карактерише се стадијумским током болести: прво, латентна фаза или скривени клинички симптоми, који се манифестују минималним променама у уринарном синдрому, затим долази до постепене декомпензације процеса са смањењем бубрежне функције са манифестним клиничким симптомима (интоксикација, астенија, заостајање у развоју, анемија). Клинички симптоми се обично јављају без обзира на слојевитост инфламаторне реакције.
Наследни нефритис може се манифестовати у различитим старосним периодима, што зависи од деловања гена, који је у потиснутом стању до одређеног времена.
Класификација
Постоје три врсте наследног нефритиса
- Опција I - клинички се манифестује као нефритис са хематуријом, губитком слуха и оштећењем ока. Ток нефритиса је прогресиван са развојем хроничне бубрежне инсуфицијенције. Тип наслеђивања је доминантан, повезан са X хромозомом. Морфолошки се открива повреда структуре базалне мембране, њено проређивање и цепање.
- Опција II - клинички се манифестује као нефритис са хематуријом без губитка слуха. Ток нефритиса је прогресиван са развојем хроничне бубрежне инсуфицијенције. Тип наслеђивања је доминантан, повезан са X хромозомом. Морфолошки се детектује истањивање базалне мембране гломеруларних капилара (посебно ламинаденсе).
- Опција III - бенигна фамилијарна хематурија. Ток је повољан, хронична бубрежна инсуфицијенција се не развија. Тип наслеђивања је аутозомно доминантан или аутозомно рецесиван. Код аутозомно рецесивног типа наслеђивања, код жена се примећује тежи ток болести.
Дијагноза Алпортовог синдрома
Предлажу се следећи критеријуми:
- присуство најмање два пацијента са нефропатијом у свакој породици;
- хематурија као водећи симптом нефропатије код испитаника;
- присуство губитка слуха код најмање једног члана породице;
- развој хроничне бубрежне инсуфицијенције код једног или више рођака.
У дијагностици различитих наследних и урођених болести, велико место се посвећује свеобухватном приступу испитивању и, пре свега, обраћању пажње на податке добијене приликом састављања родослова детета. Дијагноза Алпортовог синдрома се сматра валидном у случајевима када се код пацијента открију 3 од 4 типична знака: присуство хематурије и хроничне бубрежне инсуфицијенције у породици, присуство неуросензорног губитка слуха, патологија вида код пацијента, откривање знакова цепања гломеруларне базалне мембране са променом њене дебљине и неравним контурама током електронско-микроскопских карактеристика биопсије.
Преглед пацијента треба да обухвати клиничке и генетске методе истраживања; циљано проучавање историје болести; општи преглед пацијента узимајући у обзир дијагностички значајне критеријуме. У фази компензације, патологија се може открити само фокусирањем на синдроме као што су присуство наследног терета, хипотензија, вишеструке стигме дисембриогенезе, промене у уринарном синдрому. У фази декомпензације могу се појавити екстраренални симптоми, као што су тешка интоксикација, астенија, заостали физички развој, анемија, који се манифестују и интензивирају постепеним смањењем бубрежне функције. Код већине пацијената, са смањењем бубрежне функције, примећује се следеће: смањена ацидо- и аминогенеза; 50% пацијената примећује значајно смањење секреторне функције бубрега; ограничен опсег флуктуација оптичке густине урина; поремећај ритма филтрације, а затим смањење гломеруларне филтрације. Стадијум хроничне бубрежне инсуфицијенције се дијагностикује када пацијенти имају повишен ниво урее у крвном серуму (више од 0,35 г/л) током 3-6 месеци или дуже, и смањење гломеруларне филтрације на 25% норме.
Диференцијална дијагностика наследног нефритиса треба се првенствено спроводити са хематуричним обликом стеченог гломерулонефритиса. Стечени гломерулонефритис најчешће има акутни почетак, период од 2-3 недеље након инфекције, екстрареналне знаке, укључујући хипертензију од првих дана (код наследног нефритиса, напротив, хипотензија), смањену гломеруларну филтрацију на почетку болести, без оштећења парцијалних тубуларних функција, док су код наследног оне присутне. Стечени гломерулонефритис се јавља са израженијом хематуријом и протеинуријом, са повећаном седиментацијом еритроцита (СЕ). Типичне промене у гломеруларној базалној мембрани, карактеристичне за наследни нефритис, од дијагностичке су вредности.
Диференцијална дијагностика од дисметаболичке нефропатије спроводи се са хроничном бубрежном инсуфицијенцијом, у породици клинички откривеним хетерогеним болестима бубрега, и може постојати спектар нефропатије од пијелонефритиса до уролитијазе. Деца се често жале на бол у стомаку и периодично током мокрења, у седименту урину - оксалати.
Ако се сумња на наследни нефритис, пацијента треба упутити у специјализовано нефролошко одељење ради разјашњења дијагнозе.
Шта треба испитати?
Како испитивати?
Који су тестови потребни?
Кога треба контактирати?
Лечење Алпортовог синдрома
Режим укључује ограничења у вези са тешким физичким напорима и боравком на свежем ваздуху. Исхрана је потпуна, са довољним нивоом комплетних протеина, масти и угљених хидрата, узимајући у обзир функцију бубрега. Од великог значаја је откривање и лечење хроничних жаришта инфекције. Користе се следећи лекови: АТП, кокарбоксилаза, пиридоксин (до 50 мг/дан), карнитин хлорид. Курсеви се примењују 2-3 пута годишње. За хематурију се прописује биљни лек - коприва, сок од ароније, хајдучка трава.
У страној и домаћој литератури постоје извештаји о лечењу преднизолоном и употреби цитостатика. Међутим, тешко је проценити ефекат.
Код хроничне бубрежне инсуфицијенције користе се хемодијализа и трансплантација бубрега.
Не постоје методе специфичне (ефикасне патогенетске) терапије за наследни нефритис. Све мере лечења усмерене су на спречавање и успоравање пада функције бубрега.
Исхрана треба да буде уравнотежена и висококалорична, узимајући у обзир функционално стање бубрега. У одсуству функционалних поремећаја, исхрана детета треба да садржи довољно протеина, масти и угљених хидрата. У присуству знакова бубрежне дисфункције, количина протеина, угљених хидрата, калцијума и фосфора треба да буде ограничена, што одлаже развој хроничне бубрежне инсуфицијенције.
Физичка активност треба да буде ограничена; деци се саветује да избегавају спорт.
Треба избегавати контакт са заразним пацијентима, смањити ризик од развоја акутних респираторних болести. Неопходна је санација жаришта хроничне инфекције. Превентивне вакцинације се не спроводе за децу са наследним нефритисом, вакцинација је могућа само по епидемиолошким индикацијама.
Хормонска и имуносупресивна терапија код наследног нефритиса је неефикасна. Постоје индикације о извесном позитивном ефекту (смањење протеинурије и успоравање прогресије болести) код дуготрајне вишегодишње употребе циклоспорина А и АЦЕ инхибитора.
У лечењу пацијената користе се лекови који побољшавају метаболизам:
- пиридоксин - 2-3 мг/кг/дан у 3 дозе током 4 недеље;
- кокарбоксилаза - 50 мг интрамускуларно сваког другог дана, укупно 10-15 ињекција;
- АТП - 1 мл интрамускуларно сваког другог дана, 10-15 ињекција;
- витамин А - 1000 ИЈ/годишње/дан у 1 дози током 2 недеље;
- Витамин Е - 1 мг/кг/дан у 1 дози током 2 недеље.
Ова врста терапије помаже у побољшању општег стања пацијената, смањењу тубуларних дисфункција и спроводи се у курсевима 3 пута годишње.
Левамизол се може користити као имуномодулатор - 2 мг/кг/дан 2-3 пута недељно са паузама између доза од 3-4 дана.
Према истраживачким подацима, хипербарична оксигенација позитивно утиче на тежину хематурије и бубрежне дисфункције.
Најефикаснија метода лечења наследног нефритиса је благовремена трансплантација бубрега. У овом случају, не долази до рецидива болести код трансплантата; у малом проценту случајева (око 5%), нефритис се може развити у трансплантираном бубрегу повезан са антигенима гломеруларне базалне мембране.
Перспективни правац је пренатална дијагностика и терапија генетским инжењерингом. Експерименти на животињама показују високу ефикасност преношења нормалних гена одговорних за синтезу алфа ланаца колагена типа IV у бубрежно ткиво, након чега се примећује синтеза нормалних колагенских структура.
Прогноза
Прогноза за наследни нефритис је увек озбиљна.
Прогностички неповољни критеријуми за ток наследног нефритиса су:
- мушки пол;
- рани развој хроничне бубрежне инсуфицијенције код чланова породице;
- протеинурија (више од 1 г/дан);
- задебљање гломеруларних базалних мембрана према микроскопији;
- акустични неуритис;
- делеција у гену Col4A5.
Прогноза за бенигну фамилијарну хематурију је повољнија.