
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Лисенцефалија мозга
Медицински стручњак за чланак

Последње прегледано: 04.07.2025
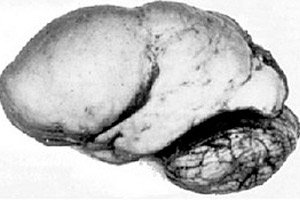
Међу органским церебралним патологијама, истиче се конгенитална аномалија развоја мозга као што је лисенцефалија, чија суштина лежи у готово глаткој површини кортекса његових хемисфера - са недовољним бројем вијуга и бразди. [ 1 ]
У потпуном одсуству конволуција дефинише се агирија, а присуство неколико широких равних конволуција назива се пахигирија. Ови дефекти, као и неке друге редукционе деформације мозга, имају код Q04.3 у МКБ-10.
Епидемиологија
Према статистици о ретким болестима, постоји 1-1,2 случаја лисенцефалије на 100 хиљада новорођенчади. [ 2 ], [ 3 ]
Према неким подацима, до 25-30% случајева класичне лисенцефалије примећује се код деце са Милеровим-Дикеровим синдромом; тачкасте мутације и делеције гена LIS1 и DCX откривају се код скоро 85% пацијената. [ 4 ]
Генетске студије 17 гена повезаних са лисенцефалијом показале су да је мутација или делеција LIS1 одговорна за 40% пацијената, а 23% је повезано са мутацијом DCX, затим следе TUBA1A (5%) и DYNC1H1 (3%).[ 5 ]
Узроци лисенцефалија
Сви познати узроци формирања мождане коре (cortex cerebri) готово или потпуно без вијуга и бразди, који повећавају „радну површину“ људског мозга и обезбеђују „продуктивност“ централног нервног система, повезани су са поремећајима у његовом перинаталном развоју. То јест, лисенцефалија се развија код фетуса. [ 6 ]
Неуспех у формирању слојева мождане коре фетуса код лисенцефалије је резултат абнормалне миграције неурона који је формирају или превременог прекида овог процеса.
Овај процес, који је неопходан за цереброкортикалну хистогенезу, одвија се у неколико фаза од 7. до 18. недеље трудноће. И, с обзиром на повећану осетљивост на генетске мутације, као и на различите негативне физичке, хемијске и биолошке утицаје, свако одступање од норме може довести до нетачне локализације неурона са могућим формирањем задебљалог слоја сиве масе кортекса без карактеристичне структуре. [ 7 ]
У неким случајевима, лисенцефалија код деце је повезана са Милеровим-Дикеровим, Вокер-Варбурговим или Норман-Робертовим синдромима.
Прочитајте такође – Развојни дефекти мозга
Фактори ризика
Поред мутација у неким генима, фактори ризика за рођење детета са тако тешким дефектом укључују кисеоничко гладовање (хипоксију) фетуса; недовољно снабдевање мозга крвљу (хипоперфузију); акутни цереброваскуларни инцидент у облику перинаталног можданог удара; патологије плаценте; вирусне инфекције труднице (укључујући TORCH); [ 8 ] проблеме са општим метаболизмом и функцијом штитне жлезде; пушење, алкохол, психотропне и наркотичке супстанце; употребу низа лекова; повећан ниво зрачења. [ 9 ]
Патогенеза
Нису сви случајеви лисенцефалије узроковани хромозомским абнормалностима и генским мутацијама. Али познати су неки гени који кодирају протеине који играју важну улогу у правилном кретању неуробласта и неурона дуж радијалних глијских ћелија - да би се формирао мождани кортекс. А мутације ових гена доводе до ове патологије. [ 10 ]
Конкретно, то су спорадичне мутације (без наслеђа) гена LIS1 на хромозому 17, који регулише цитоплазматски моторни протеин микротубула динеин, као и ген DCX на X хромозому, који кодира протеин дупликортин (лисенцефалин-X). [ 11 ]. У првом случају, стручњаци дефинишу класичну лисенцефалију (тип I), у другом – X-везану. [ 12 ]
Када се ген FLN1, који кодира фосфопротеин филамин 1, избрише, процес усмерене неуронске миграције можда уопште неће почети, што доводи до потпуног одсуства конволуција (агирије). [ 13 ]
Мутације су идентификоване у гену CDK5, који кодира ензим киназу – катализатор за интрацелуларни метаболизам, регулишући ћелијски циклус у неуронима ЦНС-а и обезбеђујући њихову нормалну миграцију током пренаталног формирања можданих структура.
Абнормалне промене у RELN гену на хромозому 7 које узрокују кортикалне гиралне дефекте код Норман-Робертсовог синдрома доводе до недостатка екстрацелуларног гликопротеина реелина, који је неопходан за регулацију миграције и позиционирања неуралних матичних ћелија током развоја кортекса церебри.[ 14 ], [ 15 ], [ 16 ]
Ген ARX кодира неаристаленски хомеобокс протеин, транскрипциони фактор који игра важну улогу у предњем мозгу и другим ткивима.[ 17 ] Деца са ARX мутацијом имају друге симптоме, као што су недостајући делови мозга (агенеза corpus callosum), абнормални гениталије и тешка епилепсија.[ 18 ],[ 19 ]
Неколико гена је повезано са лисенцефалијом. Ови гени укључују VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A и CDK5.[ 20 ]
Цитомегаловирус (ЦМВ) је повезан са развојем лисенцефалије због смањеног снабдевања мозга фетуса крвљу. Тежина ЦМВ инфекције зависи од гестацијске старости. Рана инфекција ће вероватније изазвати лисенцефалију јер се миграција неурона дешава рано у трудноћи.[ 21 ]
Поред тога, механизам настанка ове аномалије укључује непотпун или каснији прекид миграције неурона из перивентрикуларне генеративне зоне у мождану кору. И у таквим случајевима се развија или непотпуна лисенцефалија или пахигирија, у којој се формира неколико широких жлебова и конволуција (али већина њих је одсутна).
Симптоми лисенцефалија
Први знаци ове патологије (у одсуству претходно поменутих синдрома) можда се неће појавити одмах након рођења, већ након једног и по до два месеца. А најчешће се примећују следећи клинички симптоми лисенцефалије:
- мишићна хипотонија, често комбинована са спастичном парализом;
- конвулзије и генерализовани тонично-клонични напади (у облику опистотонуса);
- тешка ментална ретардација и заостајање у расту;
- оштећење неуролошких и моторичких функција.
Проблеми са гутањем узрокују потешкоће у храњењу бебе. [ 22 ]
Висок степен неуромоторног оштећења често се манифестује као тетраплегија – парализа свих удова. Могућа је деформација шака, прстију на рукама или ногама.
Код Норман-Робертовог синдрома са лисенцефалијом типа I, примећују се краниофацијалне аномалије: тешка микроцефалија, низак нагиб чела и истурени широки носни мост, широко постављене очи (хипертерлоризам), неразвијеност вилица (микрогнатија). [ 23 ]
Милеров-Дикеров синдром може се такође карактерисати абнормално малом величином главе са широким, високим челом и кратким носом, удубљењима у слепоочницама (битемпоралним удубљењима) и ниско постављеним, деформисаним ушима.
Тешки лисенцефалијски синдром карактерише микроцефалија, смањење величине очних јабучица (микрофталмија), у комбинацији са дисплазијом мрежњаче, опструктивним хидроцефалусом и одсутним или хипопластичним корпусним калозумом.
Компликације и посљедице
Међу компликацијама ове аномалије, стручњаци наводе кршење функције гутања (дисфагија) и гастроезофагеални рефлукс; рефракторну (неконтролисану) епилепсију; честе инфекције горњих дисајних путева; упалу плућа (укључујући хроничну аспирацију).
Одојчад са лисенцефалијом могу имати конгениталне органске срчане проблеме у облику дефекта атријалног септума или сложеног срчаног дефекта са цијанозом (тетралогија Фало). [ 24 ]
Последице постнаталног успоравања раста у већини случајева резултирају смрћу у року од 24 месеца након рођења.
Дијагностика лисенцефалија
Дијагноза почиње физичким прегледом детета, проучавањем медицинске историје родитеља и историје трудноће и порођаја.
Током трудноће може бити потребно тестирање феталне ДНК без ћелија, амниоцентеза или узимање биопсије хорионских ресица. [ 25 ] За више информација, погледајте – Пренатална дијагноза конгениталних болести
Инструментална дијагностика се користи за визуелизацију можданих структура и процену њихових функција:
- компјутерска томографија мозга;
- магнетна резонанца (МРИ) мозга;
- електроенцефалограм (ЕЕГ). [ 26 ]
Током трудноће, лисенцефалија на ултразвуку фетуса након 20-21 недеље може се посумњати у одсуству паријето-окципиталних и калкарних жлебова и аномалије Силвијеве фисуре мозга.
Диференцијална дијагноза
Спроводи се диференцијална дијагностика са другим синдромима конгениталних церебралних мана.
Постоји више од 20 врста лисенцефалије, од којих већина спада у 2 главне категорије: класична лисенцефалија (тип 1) и лисенцефалија типа „калдрма“ (тип 2). Свака категорија има сличне клиничке манифестације, али различите генетске мутације.[ 27 ]
Преглед мозга код лисенцефалије типа I показује мождану кору са четири слоја уместо шест, колико се види код нормалних пацијената, док је код лисенцефалије типа 2 мождана кора неорганизована и делује грудвасто или нодуларно због потпуног померања мождане коре кластерима кортикалних неурона раздвојених глиомезенхималним ткивом. Пацијенти су такође имали абнормалности мишића и очију.
- Класична лисенцефалија (тип 1):
- LIS1: Изолована лисенцефалија и Милеров-Дикеров синдром (лисенцефалија повезана са фацијалним дисморфизмом). [ 28 ]
- LISX1: мутација гена DCX. У поређењу са лисенцефалијом изазваном LIS1 мутацијама, DCX показује шестослојни кортекс уместо четири.
- Изолована лисенцефалија без других познатих генетских дефеката
- Калдрмисана лисенцефалија (тип 2):
- Вокер-Варбургов синдром
- Фукујамин синдром
- Болест мишића, очију и мозга
- Остале врсте се не могу сврстати ни у једну од две горе наведене групе:
- ЛИС2: Норман-Робертов синдром, сличан лисенцефалији типа I или Милеровом-Дикеровом синдрому, али без делеције хромозома 17.
- ЛИС3
- LISX2
Микролисенцефалија: Ово је комбинација одсуства нормалног кортикалног савијања и абнормално мале главе. Деца са правилном лисенцефалијом имају нормалне величине главе при рођењу. Деци са малом величином главе при рођењу обично се дијагностикује микролисенцефалија.
Такође је важно разликовати лисенцефалију и полимикрогирију, које су различити дефекти у развоју мозга.
Кога треба контактирати?
Третман лисенцефалија
Лисенцефалија је неизлечив органски дефект, тако да је могуће само супортивно и симптоматско лечење. [ 29 ]
Пре свега, ово је употреба антиконвулзива и антиепилептичких лекова, као и постављање гастростомске цеви у желудац (ако дете није у стању да самостално гута). Масажа је корисна.
У случајевима тешког хидроцефалуса, цереброспинална течност се уклања.
Превенција
Стручњаци препоручују будућим родитељима да потраже генетско саветовање, а труднице да се благовремено региструју код акушера и гинеколога и обаве све заказане прегледе.
Прогноза
За децу са лисенцефалијом, прогноза зависи од њеног степена, али најчешће ментални развој детета не прелази ниво од четири до пет месеци. И сва деца са овом дијагнозом пате од тешких психомоторних поремећаја и тешко лечиве епилепсије. [ 30 ]
Према NINDS-у (Амерички национални институт за неуролошке поремећаје и мождани удар), максимални животни век за особе са лисенцефалијом је око 10 година.