
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Т-ћелијски лимфоми коже
Медицински стручњак за чланак

Последње прегледано: 04.07.2025
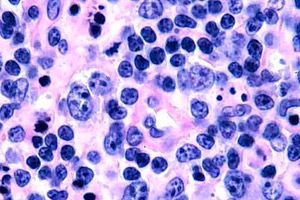
Најчешће се Т-ћелијски лимфоми региструју код старијих особа, мада се изоловани случајеви болести примећују чак и код деце. Мушкарци оболевају двоструко чешће од жена. Т-ћелијски лимфоми су епидермотропне природе.
Узроци Т-ћелијски лимфоми коже
Узроци и патогенеза кожних Т-ћелијских лимфома нису у потпуности схваћени. Тренутно, већина истраживача сматра вирус хумане Т-ћелијске леукемије тип 1 (HTLV-1) I главним етиолошким фактором који покреће развој малигних Т-ћелијских лимфома коже. Уз ово, разматра се улога других вируса у развоју Т-ћелијског лимфома: Епштајн-Баров вирус, херпес симплекс тип 6. Код пацијената са Т-ћелијским лимфомом, вируси се налазе у кожи, периферној крви и Лангерхансовим ћелијама. Антитела на HTLV-I су откривена код многих пацијената са микозис фунгоидес.
Важно место у патогенези Т-ћелијских лимфома играју имунопатолошки процеси у кожи, од којих је главни неконтролисана пролиферација клонских лимфоцита.
Цитокини које производе лимфоцити, епителне ћелије и ћелије макрофагног система имају проинфламаторне и пролиферативне ефекте (ИЛ-1, одговоран за диференцијацију лимфоцита; ИЛ-2 - фактор раста Т-ћелија; ИЛ-4 и ИЛ-5, повећавајући прилив еозинофила у лезију и њихову активацију, итд.). Као резултат прилива Т-лимфоцита у лезију, формирају се Потјерови микроапсцеси. Истовремено са повећањем пролиферације лимфоцита, сузбија се активност ћелија антитуморске одбране: природних убица, лимфоцитотоксичних лимфоцита, дендритичних ћелија, посебно Лангерхансових ћелија, као и цитокина (ИЛ-7, ИЛ-15, итд.) - инхибитора раста тумора. Улога наследних фактора се не може искључити. Присуство породичних случајева, честа детекција неких антигена хистокомпатибилности (HLA B-5 и HLA B-35 - код високо малигних лимфома коже, HLA A-10 - код мање агресивних лимфома, HLA B-8 - код еритродермичког облика микозис фунгоидес) потврђују наследну природу дерматозе.
Клиничка посматрања указују на могућу трансформацију дуготрајних хроничних дерматоза (неуродерматитис, атопијски дерматитис, псоријаза итд.) у микозис фунгоидес. Кључни фактор је дуготрајна перзистенција лимфоцита у жаришту упале, који ремете имуни надзор и подстичу настанак клона малигних лимфоцита и, тиме, развој малигног пролиферативног процеса.
Утицај физичких фактора на организам, као што су инсолација, јонизујуће зрачење и хемијске супстанце, може довести до појаве клона „генотрауматских“ лимфоцита који имају мутагени ефекат на лимфоидне ћелије и развој малигнитета лимфоцита.
Стога се Т-ћелијски лимфоми могу сматрати мултифакторијалном болешћу која почиње активацијом лимфоцита под утицајем различитих канцерогених, „генотравматизујућих“ фактора и појавом доминантног Т-ћелијског клона. Тежина поремећаја имуног надзора, клон малигних лимфоцита одређује клиничке манифестације (пјегави, плак или туморски елементи) Т-ћелијских лимфома.
Патогенеза
У раној фази микозе фунгоидес примећују се акантоза са широким процесима, хиперплазија и збијање базалних кератиноцита, вакуоларна дегенерација неких базалних ћелија, атипичне митозе у различитим слојевима епидермиса, епидермотропизам инфилтрата са продором лимфоцита у епидермис. У дермису се око крвних судова примећују мали инфилтрати, који се састоје од појединачних мононуклеарних ћелија са хиперхромним једрима - „микотичке“ ћелије. У другој фази примећује се повећање изражености дермалног инфилтрата и епидермотропизам ћелија инфилтрата, услед чега малигни лимфоцити продиру у епидермис, формирајући кластере у облику Потријеових микроапсцеса. У трећој, туморској фази, примећују се масивна акантоза и мања атрофија епидермиса, као и повећана инфилтрација епидермиса туморским лимфоцитима, који формирају вишеструке Потријеове микроапсцесе. Масивни инфилтрат се налази по целој дебљини дермиса и покрива део хиподермиса. Примећују се бластни облици лимфоцита.
Кожни велики анапластични Т-ћелијски лимфом
Представља га група лимфопролиферативних процеса које карактерише присуство пролиферата из атипичних клонских великих анапластичних CD30+ Т ћелија. По правилу, развија се секундарно у стадијуму тумора микозе фунгоидес или код Сезаријевог синдрома, али се може развити самостално или са дисеминацијом системских лимфома овог типа. Клинички, такви лимфоми одговарају такозваном декапитованом облику микозе фунгоидес у облику појединачних или вишеструких чворова, обично груписаних.
Хистолошки, пролиферат заузима скоро цео дермис са или без епидермотропизма у случају епидермалне атрофије.
Цитолошки, туморске ћелије могу варирати по величини и облику. На основу ових својстава, прави се разлика између плеоморфних Т-ћелијских лимфома средњих и великих ћелија са језгрима различитих неправилних конфигурација - увијених, вишережњевих, са густим хроматином, добро дефинисаним нуклеолусом и прилично обилном цитоплазмом; имунобластичних - са великим округлим или овалним језгрима са чистом кариоплазмом и једним централно лоцираним нуклеолусом; анапластичних - са ружним веома великим ћелијама са језгрима неправилне конфигурације и обилном цитоплазмом. Фенотипски, цела ова група припада Т-хелпер лимфомима и може бити CD30+ или CD30-.
Р. Вилемзе и др. (1994) су показали да је ток CD30+ лимфома повољнији. Генотипски се детектује клонално преуређење Т-лимфоцитног рецептора.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Симптоми Т-ћелијски лимфоми коже
Најчешћа болест у групи Т-ћелијских лимфома коже је микозис фунгоидес, која чини око 70% случајева. Постоје три клиничка облика болести: класични, еритродермични и обезглављени. Т-ћелијски лимфоми карактеришу се полиморфизмом осипа у облику мрља, плакова, тумора.
Еритродермични облик микозе фунгоидес обично почиње неконтролисаним сврабом, отоком, универзалном хиперемијом, појавом еритематозно-сквамозних лезија на кожи трупа и екстремитета, које имају тенденцију спајања и развоја еритродермије у року од 1-2 месеца. Скоро сви пацијенти имају палмарно-плантарну хиперкератозу и дифузно проређивање длака по целој кожи. Све групе лимфних чворова су знатно увећане. Увећани ингвинални, феморални, аксиларни, кубитални лимфни чворови се палпирају као „пакетићи“ густе еластичне конзистенције, нису срасли са околним ткивима, безболни. Опште стање се нагло погоршава: јавља се грозница са телесном температуром до 38-39°C, ноћно знојење, слабост и губитак тежине. Тренутно, Сезаријев синдром многи дерматолози сматрају најређом леукемијском варијантом еритродермичног облика микозе фунгоидес,
У лимфоцитограмима се примећује изражена леукоцитоза - Сезаријеве ћелије. Сезаријеве ћелије су малигне Т-помоћне ћелије, чија језгра имају пресавијену церебриформну површину са дубоким инвагинацијама нуклеарне мембране. Смртни исход се примећује након 2-5 година, а чест узрок је кардиоваскуларна патологија и интоксикација.
Декапитални облик микозис фунгоидес карактерише се брзим развојем туморских лезија на очигледно здравој кожи без претходног дуготрајног формирања плака. Овај облик карактерише висок степен малигнитета, што се сматра манифестацијом лимфосаркома. Смртоносни исход се примећује у року од годину дана.
Фазе
Класични облик микозе фунгоидес карактеришу три фазе развоја: еритематозно-сквамозни, плак и тумор.
Прва фаза подсећа на клиничку слику неких бенигних инфламаторних дерматоза - екцема, себороичног дерматитиса, плак парапсоријазе. У овој фази болести примећују се мрље различитих величина, интензивно ружичасте, ружичасто-црвене са љубичастим нијансом, округлих или овалних обриса, са релативно јасним границама, површинског мекињастог или финопластичастог љуштења. Елементи се често налазе на различитим деловима коже, најчешће на трупу и лицу. Постепено се њихов број повећава. Временом, процес може попримити карактер еритродерме (еритродермијски стадијум). Осип може постојати годинама или спонтано нестати. За разлику од бенигних инфламаторних дерматоза, елементи осипа и свраба у овој фази су отпорни на терапију.
Инфилтративно-плак фаза се развија током неколико година. На месту претходно постојећих пегавих осипа, појављују се плакови округлих или неправилних обриса, интензивно љубичасте боје, јасно разграничени од здраве коже, густи, са љускавом површином. Њихова конзистенција подсећа на „дебели картон“. Неке од њих се спонтано повлаче, остављајући подручја тамносмеђе хиперпигментације и/или атрофије (поикилодерма). Свраб у овој фази је још интензивнији и болнији, примећује се грозница и губитак тежине. У овој фази се може приметити лимфаденопатија.
У трећем, туморском стадијуму, појављују се безболни тумори густе, еластичне конзистенције, жуто-црвене боје, који се развијају из плакова или настају на очигледно здравој кожи. Облик тумора је сферног или спљоштеног облика, често подсећа на шешир печурке. Тумори се могу појавити било где. Њихов број варира од једног до десетина, величине - од 1 до 20 цм у пречнику. Када се дуго постојећи тумори распадају, формирају се чиреви са неравним ивицама и дубоким дном, који досежу до фасције или кости. Најчешће су погођени лимфни чворови, слезина, јетра и плућа. Опште стање се погоршава, јављају се и повећавају симптоми интоксикације, развија се слабост. Просечан животни век пацијената са класичним обликом микозе гљивичне од тренутка дијагнозе је од 5 до 10 година. Морталитет се обично примећује од интеркурентних болести: упале плућа, кардиоваскуларне инсуфицијенције, амилоидозе. Субјективно се осећа свраб, а када се тумори распадају, бол у погођеним подручјима.
Шта треба испитати?
Како испитивати?
Третман Т-ћелијски лимфоми коже
У еритематозно-сквамозној фази, пацијентима није потребна антитуморска терапија; прописују им се локални кортикостероиди (преднизолон, бетаметазон, деривати дексаметазона), интерферон алфа (3 милиона ИЈ дневно, затим 3 пута недељно током 3-6 месеци у зависности од клиничких манифестација или ефикасности лечења), интерферон гама (100.000 ИЈ дневно током 10 дана, циклус се понавља 12-3 пута са паузом од 10 дана), ПУВА терапија или Ре-ПУВА терапија. Ефикасност ПУВА терапије заснива се на селективном формирању ковалентних унакрсних веза псоралена са ДНК у пролиферирајућим Т-хелпер ћелијама, што инхибира њихову деобу. У другој фази, поред горе поменутих средстава, користе се системски кортикостероиди (30-40 мг преднизолона дневно током 1,5-2 месеца) и цитостатици (проспедин 100 мг дневно дневно, укупно 4-5 ињекција). Комбиновање интерферона са другим методама терапије има израженији терапеутски ефекат (интерферони + ПУВА, интерферони + цитостатици, интерферони + ароматични ретиноиди).
У стадијуму тумора, главна метода је полихемотерапија. Користи се комбинација винкристина (0,5-1 мг интравенозно једном дневно, укупно 4-5 ињекција) са преднизолоном (40-60 мг дневно орално током хемотерапије), проспидином (100 мг дневно, укупно 3 г) и интерферонима. Препоручују се фотодинамичка, терапија електронским снопом и фотофереза (екстракорпорална фотохемотерапија).