
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Ксеродерма пигментосум
Медицински стручњак за чланак

Последње прегледано: 04.07.2025
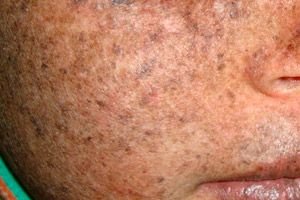
Ксеродерма пигментозум је аутозомно рецесивни генетски поремећај поправке ДНК. Болест је узрокована мутацијама у генима који су укључени у поправку ДНК оштећене ултраљубичастим зрачењем и другим токсичним супстанцама.
Најчешће, ова болест погађа децу, која се називају и „деца ноћи“. Честе компликације болести су базалноћелијски карцином и други малигни тумори коже, метастатски малигни меланом и сквамозноћелијски карцином.
 [ 1 ]
[ 1 ]
Патогенеза
Недостатак ензима УВ ендонуклеазе у ћелијама пацијента (у фибробластима) или њихово потпуно одсуство игра важну улогу у развоју болести. Овај ензим је одговоран за репродукцију ДНК оштећене ултраљубичастим зрацима. Према другим изворима, недостатак ензима ДНК полимеразе-1 игра важну улогу у развоју болести код неких пацијената. Болест се најчешће развија под утицајем зрака таласне дужине 280-310 nm. Сматра се да се пигментна ксеродерма развија услед уласка различитих фотодинамичких супстанци у организам или повећања порфирина у људском биолошком окружењу.
У раним фазама болести примећују се хиперкератоза, атрофија епидермалног жаришта, проређивање Малпигијевог слоја, повећање гранула меланина у базалном слоју ћелија и хронични инфламаторни инфилтрат у горњем слоју дермиса (углавном око крвних судова). Накнадно се откривају дегенеративне промене у колагенским и еластичним влакнима, а у фази тумора откривају се знаци карактеристични за рак коже.
Симптоми ксеродерма пигментосум
Мушкарци и жене су подједнако погођени овом болешћу. Болест почиње у раном детињству у пролеће или лето. Кожа пацијента је посебно осетљива на сунчеву светлост. Стога се први осип у облику хиперпигментације, сличне младежу, појављује на деловима коже изложеним сунчевој светлости. Осип се повећава, еритем се интензивира, постаје тамносмеђ, појављују се телангиектазије, ангиоми и атрофија. Накнадно расту папиломи и брадавице (углавном на кожи лица и врата), појављују се пеге и чиреви. Као резултат ожиљавања чира, нос постаје тањи („птичји кљун“), капак се савија. Папиломи се обично развијају у малигне туморе. Пигментирана ксеродерма, по правилу, јавља се заједно са спиноцелуларним карциномом, меланосаркомима.
Шта те мучи?
Фазе
У клиничком току ксеродерме пигментозум разликују се пет фаза: инфламаторна (еритематозна), хиперпигментација, атрофија, хиперкератоза и малигни тумори.
Током инфламаторне (еритематозне) фазе, на деловима коже изложеним сунчевој светлости (лице, врат, горњи део груди, руке, шаке) појављују се оток, црвене мрље, а понекад и пликови и везикуле. На деловима који нису изложени сунчевој светлости готово да нема осипа.
Код хиперпигментације, на месту црвених мрља појављују се светло смеђе, смеђе хиперпигментисане мрље сличне кртама.
Код атрофије, кожа се суши, постаје тања и бори се. На кожи усана и носа примећују се бројне мале, звездасте телеангиектазије и ожиљци са сјајном површином. Услед атрофије и ожиљака развијају се микротомија (смањење отвора уста), ектропион, проређивање ушију и носа, атрезија носног отвора и уста. Код 80-85% пацијената, очи су погођене: примећују се коњунктивитис, кератитис, оштећење рожњаче и слузокоже, слабљење вида. На кожи капака примећују се дисхромија, телеангиектазије, хиперкератотичке промене и тумори.
Код хиперкератозе, у горе описаним патолошким жариштима појављују се брадавичасти тумори, папиломи, кератоакантоми, фиброми, ангиофибриоми и други бенигни тумори. Стога, неки научници укључују пигментну ксеродерму у групу преканцерозних болести.
Након 10-15 година од почетка болести, малигни тумори коже (базалиом, ендотелиом, ангиосарком) се јављају у атрофично измењеним жариштима и пигментним мрљама. Тумори се уништавају за кратко време, метастазирају у унутрашње органе и доводе до смрти. У унутрашњим органима и ткивима неких пацијената примећују се опште дистрофичне промене (синдактелија другог и трећег прста на нози, зубна дистрофија, потпуни губитак косе итд.).
Обрасци
Неуролошки облик ксеродерме пигментозума манифестује се у два синдрома.
Ридов синдром карактеришу клиничке манифестације ксеродерме пигментозума и микроцефалије, идиопатија и спор раст скелетног система. У неуролошком облику болести, репарација ДНК је отежана и рендгенска терапија доводи до повећања главних патолошких жаришта.
Де Синктис-X Кокионеов синдром карактеришу следећи клинички знаци:
- развој клиничких знакова ксеродерме пигментозума на посебно фотосензитивној кожи;
- рана појава малигних тумора;
- истовремени ток спастичне парализе, микроцефалије и деменције;
- конгениталне деформације и патуљасти раст;
- неразвијеност гонада;
- чести побачаји;
- рецесивно преношење наслеђивањем.
Према неким дерматолозима, Сантис-Какионеов синдром није независна болест, већ тешка и потпуно изражена клиничка манифестација ксеродерме пигментозума.
Генетски облици
Врсте |
Џин |
Локус |
Опис |
Тип А, И, XPA |
XPA |
9q22.3 |
Ксеродерма пигментосум (XP) група А – класични облик |
Тип Б, II, XPB |
XPB |
2. квартал 21. |
XP Група Б |
Тип C, III, XPC |
XPC |
3p25 |
XP Група C |
Тип D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
КСП Група Д или Де Санцтис-Цаццхионе синдром |
Тип Е, В, XPE |
ДДБ2 |
11p12-p11 |
XP Група Е |
Тип F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Група F |
Тип Г, VII, XPG |
RAD2ERCC5 |
13q33 |
XP група G и COFS синдром (церебро-окулофациоскелетни синдром) тип 3 |
Тип V, XPV |
ПОЛХ |
6п21.1-п12 |
Варијанта ксеродерме пигментозе |
Шта треба испитати?
Како испитивати?
Кога треба контактирати?
Третман ксеродерма пигментосум
Антималаријски лекови (делагил, плаквенил, резоквин, итд.), штитећи ДНК од ултраљубичастих зрака, инхибирају процес деполимеризације, смањују осетљивост (фотосензитизацију) коже на сунчеву светлост. Ови лекови имају антиинфламаторна и хипосензибилизујућа својства. Препоручује се спровођење опште терапије заједно са витаминском терапијом (Б1, Б2, ПП, Б6, Б12, А, Е), антихистаминицима (тавегил, дифенхидрамин, супрастин), десензибилизујућим средствима (натријум тиосулфат, 10% калцијум хлорид интравенозно 10 мл).
За локални третман користе се креме и масти за сунчање.
У туморском облику пигментне ксеродерме користе се хируршке методе, течни азот и ласерски зраци. Да бисте се заштитили од сунчеве светлости, морате носити широку одећу, шешире за сунце и рукавице.
Прогноза
Већина пацијената (2/3) умире пре него што напуне 15 година. Неки пацијенти могу живети до 40-50 година.
[ 20 ]