
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Ангелманов синдром код деце и одраслих
Медицински стручњак за чланак

Последње прегледано: 04.07.2025

Постоји низ болести за које изрази попут „чувај се и нећеш се разболети“ звуче, у најмању руку, смешно. То су патологије код којих су неке менталне и физичке абнормалности својствене телу детета чак и пре рођења, али родитељи нису криви за то. Такве болести су узроковане мутацијама или абнормалностима у хромозомским сетовима и називају се хромозомским или генетским. Ангелманов синдром, Даунов синдром, Патауов синдром, Едвардсов синдром, Тарнеров синдром, Прадер-Вилијев синдром - ово је само део генетских болести са прилично пристојне листе.
Синдром срећног човека
Овог пута ћемо говорити о патологији која је добила име по енглеском педијатру Харију Енђелману, који је први покренуо питање овог проблема 1965. године, сусревши се дан раније у својој пракси са троје необичне деце, уједињене заједничким специфичним симптомима. Доктор је ову децу назвао децом луткама и написао чланак о њима, који се у почетку звао „Деца-марионете“. Сам чланак и његов наслов написани су под утиском слике виђене у једном од музеја Вероне. Слика је приказивала дечака који се смеје, а звала се „Дечак лутка“. Асоцијација детета приказаног на слици са троје деце коју је Енђелман једном срео у својој пракси подстакла је педијатра да децу обједини у једну групу због болести коју су имали.
Нема ничег изненађујућег у чињеници да децу поменуту у чланку нису приметили други лекари. На крају крајева, на први поглед се чинило да имају потпуно различите болести, толико је различита била општа клиничка слика болести у 3 различита случаја. Можда би „нова“ хромозомска патологија заинтересовала друге научнике, али у то време генетика још није била довољно развијена да потврди хипотезу енглеског лекара. Стога је, након извесног интересовања за њу, чланак дуго бачен на задњу полицу.
Следеће помињање Ангелмановог синдрома, како се сада звао чланак енглеског педијатра Г. Ангелмана, датира из раних 80-их година 20. века. И тек 1987. године било је могуће пронаћи разлог зашто се мали део деце рађа са таквим одступањима да споља изгледају као да су стално насмејани и срећни. У ствари, то уопште није истина, а осмех је само гримаса, иза које се крије несрећна људска душа и бол родитеља.
Епидемиологија
Према статистици, хромозомска мутација код детета може се развити и на позадини сличних мутација код родитеља и у одсуству таквих. Не постоји јасна наследна природа Ангелмановог синдрома (АС), али је вероватноћа развоја патологије код родитеља са хромозомским мутацијама прилично висока.
Такође је занимљиво да ако породица већ има дете са АС, постоји један проценат шансе да се добије друго дете са истим поремећајем, чак и ако су родитељи здрави.
Још увек нема тачне статистике о броју пацијената са Ангелмановим синдромом. Можда је разлог разноликост симптома, који се могу јавити у одређеном саставу или се уопште не јављати дуго времена. Претпоставља се да је преваленција болести: 1 дете на 20.000 новорођенчади. Али ова бројка је веома приближна.
Узроци Ангелманов синдром
Ангелманов синдром је медицински назив за хромозомску патологију, али није ни приближно једини. Људи ову болест називају синдромом деце лутака, синдромом срећне лутке, Петрушкиним синдромом и синдромом смејуће лутке. Људи смишљају свакаква имена (понекад чак и увредљива за саме пацијенте и њихове родитеље), али болест је болест, колико год смешно изгледала и без обзира на разлоге.
А разлози за развој Ангелмановог синдрома, као и многих других генетских патологија, у свим случајевима су поремећаји у структури једног од хромозома или хромозомског скупа у целини. Али у нашем случају, цео проблем лежи у хромозому 15, пренетом од мајке. То јест, очев хромозом у овом случају нема одступања, али женски пролази кроз одређене мутације.
Према врсти хромозомске абнормалности, Ангелманов синдром се класификује као хромозомска мутација. Такве мутације се сматрају:
- Делеција (одсуство дела хромозома који садржи одређени скуп гена; ако један од гена недостаје, говоримо о микроделецији), која је резултат два прекида и једног поновног спајања, када се губи део оригиналног хромозома.
- Дупликација (присуство додатног дела хромозома који је копија постојећег), што у већини случајева доводи до смрти особе, а ређе до неплодности.
- Инверзија (окретање једног од делова хромозома за 180 степени, тј. у супротном смеру, а затим се гени у њему налазе у супротном редоследу), када се прекинути крајеви хромозома повезују редоследом који се разликује од оригиналног.
- Уметање (ако део генетског материјала у хромозому није на свом месту),
- транслокација (ако је одређени део хромозома причвршћен за други хромозом; таква мутација може бити обострана без губитка делова).
Примајући мутирани хромозом од неслутеће мајке, дете је осуђено на рођење са абнормалностима. Најчешћи узрок Ангелмановог синдрома се и даље сматра делецијом мајчиног 15. хромозома, када недостаје мали део. Мање уобичајене мутације код синдрома „лутке која се смеје“ сматрају се:
- транслокација,
- унипатернална дисомија (ако је дете добило пар хромозома од оца, мајчин хромозом је одсутан),
- мутација гена у ДНК, који су и главни грађевински (генетски) материјал и упутства за његову исправну употребу (посебно, мутација гена ube3a у мајчином хромозому).
Присуство једне од ових мутација код родитеља је фактор ризика за развој Ангелмановог синдрома код деце. Али не само хромозомске мутације, већ и геномске (које су повезане са квантитативном променом у хромозомским сетовима и чешће су од хромозомских) могу изазвати развој болести код детета. Уобичајене геномске мутације укључују хромозомску трисомију (ако хромозомски сет особе има више од 46 хромозома).
Да би се патологија појавила код детета, уопште није неопходно да родитељи имају хромозомске абнормалности. Па ипак, постоји одређени проценат пацијената чија је болест наследна.
Патогенеза
Хајде да се мало дубље позабавимо биологијом, или прецизније, генетиком. Генетске информације сваког појединачног људског организма садржане су у 23 пара хромозома. Један хромозом из пара се преноси на дете од оца, други од мајке. Сви парови хромозома се разликују по облику и величини и носе одређене информације. Тако је 23. пар хромозома (X и Y хромозоми) одговоран за формирање полних карактеристика бебе (XX - девојчица, XY - дечак, док Y хромозом дете може добити само од оца).
Идеално је да дете од родитеља добије 46 хромозома, који формирају његове генетске карактеристике, предодређујући га као јединку. Већи број хромозома назива се трисомија и сматра се одступањем од норме. На пример, присуство хромозома 47 у хромозомском сету (кариотип, одређујући врсту и индивидуалне карактеристике) узрокује појаву Дауновог синдрома.
Ако се хромозоми обоје посебном бојом, онда се под микроскопом могу видети пруге различитих нијанси дуж сваке од њих. Унутар сваке пруге налази се огроман број гена. Све ове пруге су научници нумерисали и имају фиксну локацију. Одсуство једне од пруга сматра се одступањем од норме. Код Ангелмановог синдрома, веома често се може посматрати одсуство сегмената мајчиног хромозома у интервалу q11-q13, који се налази у дугом краку, чији је број ДНК база само око 4 милиона.
Главна компонента хромозома сматра се невероватно дугим молекулом ДНК који садржи хиљаде гена и десетине и стотине милиона азотних база. Тако, хромозом 15, одговоран за развој Ангелмановог синдрома и неколико других, садржи 1200 гена и око 100 милиона база. Било какви поремећаји у структури молекула ДНК ће сигурно утицати на изглед и развој будућег детета.
Генетске информације садржане у генима се претварају у протеин или РНК. Овај процес се назива експресија гена. На овај начин, генетске информације примљене од родитеља добијају и облик и садржај, што је отелотворено у њиховом јединственом женском или мушком наследнику.
Постоји низ патологија са некласичним типом наслеђивања, укључујући Ангелманов синдром, код којих гени примљени од родитеља као део упарених хромозома носе јединствени отисак родитеља и манифестују се на различите начине.
Дакле, Ангелманов синдром је упечатљив пример геномског импринтинга, код кога је експресија гена у телу детета директно зависна од тога од ког родитеља су примљени алели (различити облици једног гена, примљени од оца и мајке, смештени на идентичним деловима упарених хромозома). То јест, само аномалије у мајчином хромозому доводе до развоја синдрома, док мутације и структурни поремећаји очевог хромозома узрокују потпуно различите патологије.
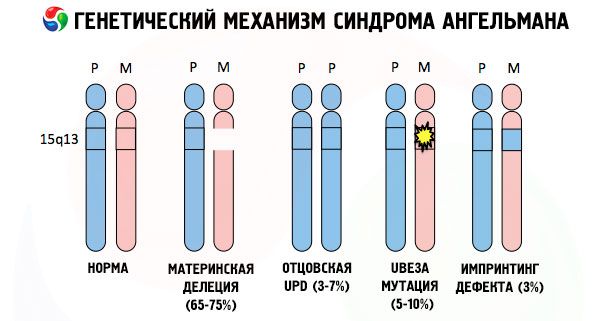
Код ове патологије долази до недостатка одређених гена у мајчином хромозому или губитка/смањења активности појединачних гена (у великој већини случајева, гена ube3a, који је укључен у метаболизам убиквитина, протеина који регулише разградњу других протеина). Као резултат тога, детету се дијагностикују менталне развојне абнормалности и физичке деформације.
Симптоми Ангелманов синдром
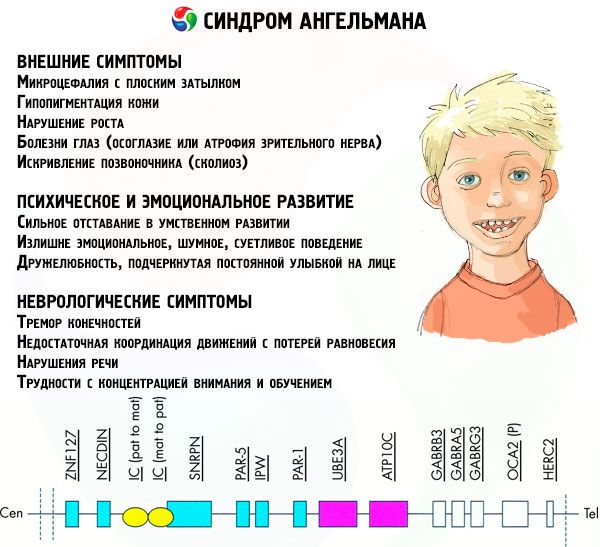
Симптоми Ангелмановог синдрома утичу на различите аспекте живота и развоја детета: физичке, неуролошке, менталне. На основу тога, могу се идентификовати 3 групе симптома које указују на развој ове патологије.
- Спољашњи или физички симптоми:
- несразмерно мала глава у поређењу са телом и удовима, који су нормалне величине,
- преширока уста,
- скоро увек је осмех на лицу (са отвореним устима),
- ретки зуби,
- уска горња усна,
- често испупчен широк језик,
- избочена доња вилица,
- зашиљена брада,
- веома светла кожа, често коса (албинизам, повезан са чињеницом да тело не производи пигмент меланин),
- тамне мрље на светлој кожи (хипопигментација због недовољне производње меланина)
- физички или спољашњи симптоми: очне болести као што су страбизам или атрофија очног живца,
- закривљеност кичме (сколиоза),
- укочене ноге (приликом ходања, особа не савија ноге у коленима због мале покретљивости зглобова, па отуда и поређење са ходом лутке).
- Симптоми повезани са менталним и емоционалним развојем:
- тешка ментална ретардација,
- претерано емотивно, бучно, нервозно понашање,
- често пљескање рукама,
- изражена љубазност, наглашена сталним осмехом на лицу,
- чест смех без разлога.
- Неуролошки симптоми:
- тремор удова,
- недовољна координација покрета са губитком равнотеже,
- смањен мишићни тонус,
- разни поремећаји спавања,
- чести хистерични напади у детињству,
- поремећаји говора (дете почиње касно да говори, има лоше комуникацијске вештине и нејасан говор),
- хиперактивност на позадини повећане раздражљивости,
- тешкоће са концентрацијом и учењем.
Али ово је генерализована слика болести. У ствари, клиничка слика Ангелмановог синдрома у великој мери зависи од стадијума развоја болести и врсте хромозомске мутације која је изазвала патологију. То значи да се симптоми болести могу значајно разликовати код различитих пацијената, што нам дуго није дозвољавало да разликујемо патологију од других са сличном клиничком сликом.
Међу укупним бројем симптома, можемо издвојити оне који су карактеристични за све пацијенте без изузетка:
- тешка ментална ретардација,
- непримерено понашање (неразумно смејање, повећана раздражљивост, лоша концентрација, стање еуфорије),
- неразвијеност моторичких способности,
- лоша координација покрета, атаксија хода (неуједначен темпо, љуљање са стране на страну, итд.), тремор удова.
- поремећај развоја говора са превлашћу невербалних средстава комуникације.
Међу симптомима које примећује велика већина пацијената, могу се разликовати следећи:
- несразмерност између главе и тела узрокована одложеним физичким развојем,
- код многих пацијената облик лобање је такав да величина мозга остаје мања него код здравих људи (микроцефалија),
- епилептични напади пре 3 године старости са прогресивним смањењем јачине и учесталости у старијем узрасту,
- изобличење ЕЕГ параметара (флуктуације и висока амплитуда нискофреквентних таласа).
Ови симптоми су прилично чести, међутим, 20% пацијената са Ангелмановим синдромом их нема.
Још ређе је могуће дијагностиковати такве манифестације болести као што су:
- тешки или благи страбизам,
- лоша контрола покрета језика, што доводи до тога да пацијенти често исплазују језик без икаквог разлога,
- тешкоће са гутањем и сисањем, посебно код мале деце,
- поремећај пигментације коже и очију,
- руке подигнуте или савијене током ходања,
- хиперрефлексија,
- поремећаји спавања, посебно у детињству,
- често саливирање,
- неутољива жеђ,
- претерано активни покрети жвакања,
- преосетљивост на топлоту,
- раван потиљак,
- избочена доња вилица,
- глатке дланове.
Прилично велики проценат пацијената има проблеме са мокрењем, које лоше контролишу, оштећене фине моторичке способности, што ствара тешкоће у самопомоћи и учењу, и прекомерну тежину. Готово сви пацијенти доживљавају пубертет касније од здравих вршњака.
Деца са Ангелмановим синдромом добро перципирају усмени говор и разумеју га, али не желе да учествују у разговору, ограничавајући свој говор на неколико десетина речи неопходних у свакодневном животу. Међутим, у одраслом добу такви пацијенти изгледају млађе од својих вршњака без генетских патологија.
Многи симптоми Ангелмановог синдрома су непостојани, па се клиничка слика болести значајно мења са годинама. Конвулзије и епилептични напади постају ређи или потпуно нестају, пацијент постаје мање узбуђен, а сан се побољшава.
Компликације и посљедице
Ангелманов синдром је тешка, тренутно практично неизлечива хромозомска патологија која пацијентима одузима могућност да живе нормалним животом. Какав ће бити живот детета са АС у великој мери зависи од врсте хромозомске абнормалности.
Дуплирање сегмента хромозома је у већини случајева неспојиво са животом. Чак и ако такви пацијенти не умру у детињству и достигну пубертет, немају шансе да имају децу.
Брисање или одсуство дела гена које се најчешће јавља код Ангелмановог синдрома представља препреку детету да научи да хода и говори. Таква деца имају тежи облик менталне ретардације, а епилептични напади се јављају чешће, а њихов интензитет је много већи него код пацијената са другим хромозомским абнормалностима.
Ако постоји само мутација једног гена, уз дужну пажњу и приступ детету се могу научити основе бриге о себи, комуникације и интеракције у групи, иако ће и даље заостајати за својим вршњацима у развоју.
За децу са Ангелмановим синдромом, која су по природи љубазна, најважнија је љубав и пажња родитеља. Само у том случају ће образовање детета донети плодове, чак и ако су мали. Наравно, пацијенти са АС неће моћи да уче у редовној школи. Потребни су им посебни часови где ће деца прво бити учена да се концентришу, а затим ће им се постепено давати основе школског знања.
Дијагностика Ангелманов синдром
Ангелманов синдром је конгенитална развојна патологија. Али због одређених околности, често га је немогуће дијагностиковати у одојчади и раном детињству. То је због неспецифичности и слабе изражености симптома код одојчади и деце млађе од 3 године. А преваленција болести у нашој земљи није толико велика да су лекари научили да је препознају међу њеним вршњацима.
Ангелманов синдром код одојчади може се манифестовати као смањен мишићни тонус, што се манифестује проблемима са храњењем (слабост рефлекса сисања и гутања), а касније и тешкоћама у учењу ходања (таква деца почињу да ходају много касније). Ови симптоми су први знаци развојне абнормалности код бебе, која може бити повезана са хромозомском абнормалношћу. Само генетска анализа може потврдити ову претпоставку.
Посебна пажња се посвећује деци чији родитељи имају различите геномске или хромозомске поремећаје. На крају крајева, болест се можда у почетку не манифестује, а ако се патологија открије на време, почевши интензивно радити са дететом, могуће је постићи знатно већи успех у учењу, успоравајући напредовање болести.
Ако родитељи имају различите хромозомске абнормалности, генетска анализа се спроводи чак и пре него што се беба роди, јер је СА једна од патологија које се могу открити у ембрионалној фази.
Прикупљање материјала за генетска истраживања може се обавити на два начина:
- инвазивна (са одређеним процентом ризика, јер је потребно продрети у материцу да би се узео узорак амнионске течности),
- неинвазивна (анализа ДНК бебе из мајчине крви).
Затим се спроводе следеће студије:
- флуоресцентна ин ситу хибридизација (FISH метода) – везивање ДНК сонде обележене посебном бојом за ДНК која се испитује, након чега следи испитивање под микроскопом.
- анализа мутација у гену ube3a и импринтираним генима,
- Анализа метилације ДНК коришћењем посебних метода које се користе у генетици.
Генетски тестови пружају прилично тачне информације у случају хромозомских абнормалности, што значи да будући родитељи унапред знају на шта да се припреме. Међутим, постоје изузеци. Код одређене групе пацијената, у присуству свих симптома који указују на патологију, резултати теста остају нормални. То јест, патологија се може идентификовати само пажљивим посматрањем детета од раног детињства: како једе, када је почело да хода и говори, да ли савија ноге приликом ходања итд.
Поред FISH методе, међу инструменталним дијагностичким методама за Ангелманов синдром, може се издвојити томографија (CT или MRI), која помаже у одређивању стања и величине мозга, и електроенцефалограм (EEG), који показује како функционишу поједини делови мозга.
Лекари обично постављају коначну дијагнозу у узрасту од 3-7 година, када пацијент већ има већину симптома и видљива је динамика развоја болести.
Који су тестови потребни?
Диференцијална дијагноза
Ангелманов синдром је генетска патологија која практично нема специфичне манифестације. Већина симптома може подједнако указивати и на АС и на друге генетске патологије.
Диференцијална дијагноза Ангелмановог синдрома се спроводи са следећим патологијама:
- Пит-Хопкинсов синдром (пацијенте карактерише ментална заосталост, весели карактер, осмех, имају прилично велика и широка уста, примећује се микроцефалија). Разлика је у нападима хипервентилације и задржавању даха у будном стању.
- Кристијансонов синдром (пацијенти су ментално заостале особе са веселим расположењем, неспособне да говоре, карактерише их микроцефалија, атаксија, конвулзије, невољни покрети мишића).
- Моват-Вилсонов синдром (симптоми: ментална ретардација, епилептични напади, шиљата брада, отворена уста, срећан израз лица, микроцефалија). Карактеристике: велика удаљеност између очију, очи косо увучене ка унутра, заобљен врх носа, уназад окренута ушна шкољка.
- Кабуки синдром (карактерише га блага до умерена ментална ретардација, проблеми са говором и моториком, мишићна слабост, епилептични напади, микроцефалија, дуги интервали између свраба и поремећена координација). Карактеришу га лучно подигнуте обрве, изврнут латерални део доњег капка, широко постављене очи, дугачке палпебралне фисуре са дугим, густим трепавицама.
- Ретов синдром (диференцијација од АС код жена). Симптоми: одложени развој говора, напади, микроцефалија. Разлика је у томе што нема срећног израза лица, јављају се напади апнеје и апраксије, што временом напредује.
- Аутозомно рецесивни синдром менталне тардације 38 (симптоми: изражена ментална ретардација са кашњењем у моторичким вештинама и говору, мишићна слабост, проблеми са храњењем у детињству, импулсивност). Карактеристична карактеристика је плава боја ириса.
- Синдром дупликације гена MECP 2 (диференцијација од SA код мушкараца). Симптоми: тешка ментална ретардација, мишићна слабост од детињства, проблеми са говором или недостатак говора, епилепсија. Карактеристике: прогресивна миопатија, стално понављајуће инфекције.
- Клифстрин синдром (симптоми: проблеми са говором и размишљањем, слабост мишића, поремећаји спавања, недостатак пажње, отворена уста, хиперактивност, напади, атаксија, поремећаји равнотеже). Карактеристичне особине: равно лице, кратак прљави нос, широко постављене очи, велика извучена доња усна, агресивни изливи.
- Смит-Магенисов синдром (карактерише га напади, проблеми са спавањем, поремећаји интелектуалног и моторичког развоја). Карактеристичне особине укључују широко и равно лице и истакнуто чело.
- Кулен-де Врисов синдром (блага до умерена ментална ретардација, мишићна слабост, напади, дружељубивост). Карактеристичне особине: дуго лице са високим челом, штрчеће уши, косе очи, велика покретљивост зглобова, урођене срчане мане.
- Фелан-Макдермидов синдром (симптоми: ментална ретардација, поремећаји говора или недостатак говора). Одлике: велике руке са развијеним мишићима, мишићна слабост од рођења, слабо знојење.
Патологије попут недостатка аденил сукцината, синдрома аутозомно рецесивне менталне ретардације 1, синдрома дупликације хромозома 2q23.1, синдрома хаплоинсуфицијенције гена FOXG1, STXBP1 или MEF2C и неких других могу се „похвалити“ симптомима сличним Ангелмановом синдрому.
Задатак лекара је да постави тачну дијагнозу, разликујући Ангелманов синдром од патологија са сличним симптомима, и пропише ефикасан третман који је релевантан за дијагностиковану фазу болести.
Третман Ангелманов синдром
Ангелманов синдром је једна од оних патологија за које медицина још увек трага за ефикасним лечењем. Етиолошки третман болести је у фази развоја различитих метода и средстава, од којих многа још нису тестирана на људима. То значи да се за сада лекари морају ограничити на симптоматску терапију, која помаже да се некако ублажи незавидна ситуација деце и одраслих са марионетским синдромом, који пате од епилептичних напада, саливације, хипотензије и поремећаја спавања.
Дакле, могуће је смањити учесталост и јачину епилептичних напада уз помоћ правилно одабраног антиконвулзивног лека. Али цела тешкоћа је у томе што се напади код пацијената са СА разликују од обичних епилептичних напада по томе што их карактерише неколико врста напада, што значи да се стање може ублажити давањем неколико лекова одједном.
Најпопуларнији антиконвулзиви који се користе за лечење Ангелмановог синдрома су: валпроинска киселина, топирамат, ламотригин, леветирацетам, клоназепам и лекови на њиховој бази. Мање се користе лекови на бази кармазепина, фенитоина, фенобарбитала, етосуксимида, јер неки од њих могу изазвати парадоксални ефекат који се састоји у појачавању и повећању учесталости епилептичних напада. То се дешава ако се лек користи као део монотерапије.
За лечење слињења обично се користе две методе: медикаментозна (лекови који сузбијају производњу пљувачке) и хируршка, која подразумева реимплантацију пљувачних канала. Али у случају саркоме шупљине, ове методе се сматрају неефикасним и питање остаје отворено. Родитељи и они који брину о таквим пацијентима морају обратити посебну пажњу на ово питање, јер сами пацијенти обично не контролишу слињење, а неки једноставно нису у стању да се сами брину о себи.
Још један проблем је кратко трајање сна. Често деца са Ангелмановим синдромом спавају не више од 5 сати, што негативно утиче на функционисање целог тела. Лако узбудљива, активна деца која воле игре и комуникацију (чак и ако покушавају да се ограниче на невербалне методе) приметно су уморна током дана. Да би се добро одморило, телу је потребан дубок, пун сан, али управо у томе је цака.
Чини се да би седативни лекови (фенотиазини и атипични антипсихотици) који смирују нервни систем требало да буду довољни да побољшају сан код узбудљивих пацијената. Али у случају АС, употреба таквих лекова је препуна појаве негативних ефеката. Стога, лекари и даље преферирају благе таблете за спавање, као што су Мелатонин (природни хормонски лек на бази хормона спавања), који се пацијентима даје сат времена пре спавања у количини од 1 таблете, и Дифенхидрамин. Учесталост примене и дозирање одређује лекар у зависности од стања и старости пацијента.
Понекад пацијенти са Ангелмановим синдромом имају проблема са варењем и столицом. Столицу можете побољшати лаксативима (пожељно биљним).
Или можете приступити проблему другачије, као што су то урадили амерички лекари, на основу неких метода лечења аутизма, јер су многи симптоми карактеристични за АС такође карактеристични за аутизам (импулсивност, невољни покрети, понављајуће радње, дефицит пажње, проблеми у комуникацији итд.). Примећено је да уношење хормона секретина, који нормализује варење и столицу, позитивно утиче на пажњу пацијената, а окситоцин помаже у побољшању когнитивних способности и памћења детета, и исправљању понашања.
Истина, само хормони нису довољни, посебно када су у питању деца. Код Ангелмановог синдрома индикована је бихејвиорална терапија, рад са психологом и логопедом (подучавање методама невербалне комуникације и знаковног језика). Образовање такве деце треба да се заснива на индивидуалном програму уз учешће специјално обучених наставника, психолога и родитеља. Нажалост, то није свуда могуће, а породице остају саме са својим проблемом.
Пошто многи млади пацијенти са АС пате од ниског мишићног тонуса и проблема са зглобовима, велика пажња се посвећује физиотерапији. Најчешће, лекари прибегавају употреби парафинских апликација, електрофорезе и магнетне терапије.
Активна тоничка масажа и посебне вежбе терапијске физичке обуке помоћи ће болесном детету да после неког времена стане на ноге и самоуверено хода. У том погледу је посебно корисна аквагимнастика, која се препоручује код СА у хладној води. Повећава тонус мишића и учи дете да контролише своје тело и координира покрете.
Антиконвулзивни третман
Најопаснији симптом Ангелмановог синдрома су напади слични онима код епилепсије. Овај симптом се примећује код 80% пацијената, што значи да свима њима треба прописати ефикасан антиконвулзивни третман.
Лечење епилептичних напада се спроводи уз помоћ витамина и антиконвулзива. Код Ангелмановог синдрома, праћеног конвулзивним синдромом, корисни ће бити витамини групе Б, као и витамини Ц, Д и Е. Али самостално прописивање витаминске терапије у овом случају је веома опасно, јер неконтролисани унос витамина може смањити ефикасност антиепилептичких лекова и изазвати нове, теже и дуже нападе.
Избор антиконвулзивних лекова и прописивање њихове ефикасне дозе такође треба да обави лекар специјалиста. Он или она такође одлучује да ли ће један лек бити довољан или ће пацијент морати да узима 2 или више лекова током дужег времена.
Већини пацијената лекари прописују лекове валпроинске киселине (Валпроинска киселина, Депакин, Конвулекс, Валпарин, итд.), који спречавају нападе и побољшавају расположење и ментално стање пацијената.
Валпроинска киселина је доступна у облику таблета, сирупа и ињекционих раствора. Најпопуларнији лек је лек са продуженим ослобађањем „Депакин“ у таблетама и као раствор за интравенозну примену. Дозу лека одређује лекар појединачно у зависности од тежине, старости и стања пацијента.
Лек се узима током оброка 2 до 3 пута дневно. Просечна дневна доза је 20-30 мг на 1 килограм тежине пацијента, максимална је 50 мг/кг дневно.
Контраиндикације за употребу. Не користити у случају дисфункције јетре и панкреаса, хеморагичне дијатезе, хепатитиса, порфирије и преосетљивости на лек.
Нежељени ефекти укључују тремор руку, поремећаје варења и столице и промене телесне тежине.
„Топирамат“ је такође лек избора за СА. Производи се у облику таблета и користи се и као део монотерапије и у комбинацији са другим лековима.
Начин примене и дозирање. Таблете узимати орално без обзира на унос хране. Почетна дневна доза за одрасле је 25-50 мг, за децу - 0,5-1 мг/кг. Сваке недеље, доза се повећава према упутствима лекара.
Лек се не сме узимати током трудноће и лактације, као и у случају преосетљивости на његове компоненте. Лек има много различитих нежељених ефеката.
Лекови које лекар може прописати за Ангелманов синдром: Кломазепам, Ривотрил, Ламотригин, Сеизар, Ламиктал, Леветирацетам, Кепра, Епитера, итд.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Традиционална медицина и хомеопатија
Традиционална медицина, попут хомеопатских препарата, је наравно релативно безбедна, али ефикасност таквог лечења Ангелмановог синдрома може се сматрати контроверзном.
Иако народни третман ипак може помоћи у неким стварима. Говоримо о заустављању епилептичних напада. У том погледу, биљни третман може бити прилично ефикасан.
Добар ефекат пружа лековита колекција на бази божура, сладића и водене леће (компоненте се узимају у једнаким количинама). Биље је потребно млевети у брашно. Након 2 недеље од почетка узимања, можете приметити значајно смањење учесталости напада.
Декорација лаванде (1 кашичица по чаши кључале воде) је такође корисна за грчеве. Смеша се кува 5 минута и инфузира пола сата. Лек се узима ноћу 14 дана.
Водена (или алкохолна) инфузија мајчине траве сматра се ефикасном код епилептичних напада.
Од хомеопатских препарата за спречавање напада код Ангелмановог синдрома, можете користити лекове на бази камилице и мајчине траве, Ацидум хидроцијаникум, Аргентум нитрикум, Калијум броматум, Арсеникум албум. Али треба узети у обзир да само хомеопатски лекар може прописати ефикасне и безбедне дозе лекова у сваком конкретном случају.
Превенција
Као што је читалац вероватно већ разумео, медицина још увек није у стању да спречи генске мутације и друге хромозомске абнормалности, међутим, као ни да исправи ситуацију. То се може десити било коме, јер се деца са Ангелмановим синдромом рађају од здравих родитеља, а генетика, која је тренутно једна од најмање проучаваних грана медицине, још увек не може ово да објасни.
Једино што се може учинити јесте одговорно приступити планирању трудноће, регистровати се и благовремено подвргнути прегледима. Али опет, таква мера ће бити више едукативна него превентивна, као и сваки преглед. Али млади родитељи ће унапред знати на шта да се припреме, а у случају позитивног одговора, одлучиће да ли могу да преузму такву одговорност као што је одгајање болесног детета.
Прогноза
Прогноза за Ангелманов синдром зависи од природе хромозомске абнормалности и благовремености њеног откривања. Најтеже су погођена она деца чији хромозом 15 садржи „празнине“ у генима (делеција). Вероватноћа да ће такви пацијенти ходати и говорити је изузетно мала. Остали случајеви се могу исправити пажљивим приступом и љубављу према свом детету.
Нажалост, такви пацијенти неће моћи да постану пуноправни чланови друштва, упркос чињеници да су далеко од глупих, разумеју говор и његово значење. Међутим, имаће проблема са комуникацијом до краја живота. Пацијенте је могуће учити знаковном језику од детињства, али их не може присилити да комуницирају речима. Речник „говорећих“ пацијената је ограничен на минимум речи које се користе у свакодневном животу (5-15 речи).
Што се тиче очекиваног животног века и општег здравственог стања пацијената са Ангелмановим синдромом, овде се бројке крећу око просечних вредности. У одраслом добу, пацијенти се углавном суочавају са здравственим проблемима као што су сколиоза и гојазност, који, уз правилан приступ лечењу, нису опасни по живот.