
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Алкаптонурија је конгенитална ензимска абнормалност
Медицински стручњак за чланак

Последње прегледано: 04.07.2025
Један од веома ретких метаболичких поремећаја, алкаптонурија, односи се на конгениталне аномалије у метаболизму аминокиселине тирозин.
Овај синдром се може назвати и недостатком хомогентизат оксидазе, хомогентизинуријом, наследном охронозом или болешћу црног урина.[ 1 ]
Епидемиологија
Према статистици, нема више од девет случајева алкаптонурије на милион људи. А у већини европских земаља постоји један случај на 100-250 хиљада живорођене деце.
Међу европским земљама, изузетак је Словачка (посебно релативно мали северозападни регион), где је преваленција алкаптонурије један случај на 19.000 новорођенчади. То је највероватније због чињенице да је међу словачким ромским породицама које тамо живе ниво инбридинга (бракова између рођака) највиши у Европи: 10-14%. [ 2 ]
Узроци алкаптонурија
Тачни узроци алкаптонурије, као конгениталног поремећаја катаболизма (метаболичког разградње) ароматичне (хомоцикличне) α-аминокиселине тирозина, су утврђени: ова врста метаболичког поремећаја је последица хомозиготних или сложених хетерозиготних мутација једног од хиљада гена на хромозому 3, тачније, HGD гена на локусу 3q21-q23 на дугом краку хромозома. Овај ген кодира нуклеотидне секвенце ензима јетре хомогентизат-1,2-диоксигеназе [ 3 ] (такође назване хомогентизинска киселинска оксидаза или хомогентизат оксидаза) - металопротеина који садржи гвожђе, неопходног за једну од фаза разградње тирозина у телу. [ 4 ], [ 5 ]
Дакле, алкаптонурија је дефект ензима хомогентизат-1,2-диоксигеназе, или прецизније, резултат његовог генетски одређеног недостатка или потпуног одсуства. [ 6 ]
Као конгенитални ензимски недостатак, алкаптонурија се наслеђује као аутозомно рецесивна особина, односно да би се алкаптонурија јавила код деце, оба родитеља морају имати модификовани ген за ензим, јер свако од њих преноси на дете само једну копију гена од две доступне.
Према најновијим подацима, постоји више од две стотине варијанти модификације HGD гена, а најчешће се примећују мисенс мутације, транслокација и сплајсинг.
Фактори ризика
Једини фактор ризика за развој ове конгениталне ензимопатије је њено присуство у породичној историји и наслеђивање две модификоване копије HGD гена, ако родитељи не показују алкаптонурију (ризик од преношења аномалије је 25%), или један од родитеља има овај поремећај. [ 7 ]
Патогенеза
Тирозин игра кључну улогу у синтези протеина, производњи хромопротеина - пигмента коже меланина, као и тироидних хормона и катехоламинских неуротрансмитера.
Механизам за регулисање количине тирозина у ћелијама је веома сложен, а тело нормализује његов вишак садржај разградњом. Процес катаболизма тирозина, као и свих ароматичних аминокиселина, је вишестепен и одвија се у неколико фаза. Свака фаза метаболичке разградње тирозина одвија се уз учешће специфичног ензима и формирање међупроизводног једињења.
Дакле, прво се аминокиселина разлаже на пара-хидроксифенилпируват, који се претвара у алкаптон – 2,5-дихидроксифенилсирћетну или хомогентизинску киселину. Затим би алкаптон требало да се трансформише у малесирћетну киселину, али се то не дешава. [ 8 ]
А патогенеза алкаптонурије састоји се у престанку биохемијских реакција катаболизма тирозина у фази формирања хомогентизинске киселине: једноставно нема ензима потребног за њено разграђивање – хомогентизат оксидазе.
Хомогентизинску киселину тело не користи и може се акумулирати излучивањем преко бубрега. Поред тога, оксидује се у бензохиноацетат (бензохинонсирћетну киселину), која, везивањем за молекуле ткива и телесних течности, формира биополимерна једињења обојена попут меланина.
Акумулација ових међупроизвода у ткиву доводи до поремећаја колагенске структуре хрскавичног ткива, што смањује његову еластичност – са појавом многих клиничких знакова алкаптонурије и развојем компликација.
Симптоми алкаптонурија
Алкаптонурија код новорођенчади и одојчади карактерише се потамњењем урина. Када је изложен ваздуху, урин на пеленама, пеленама и доњем вешу постаје тамносмеђ; то је због акумулације и ослобађања хомогентизинске киселине, која се оксидује у бензохиноацетат. [ 9 ]
У одсуству других симптома, алкаптонурија код мале деце се често не препознаје благовремено, јер урин може потамнити након неколико сати мокрења. Према неким подацима, само петина деце млађе од 12 месеци која су рођена са недостатком овог ензима идентификује се у клиничким условима. Стога је веома важно да родитељи обрате пажњу на бригу о својој одојчади.
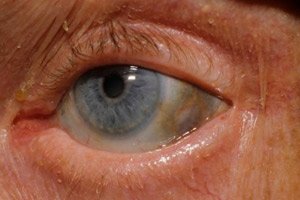
Поред тога, рани знаци укључују пигментацију (плавкасто-сиву боју) беоњаче очију и хрскавица ушију и носа, што се често назива охроноза.[ 10 ]
Временом се појављују и други симптоми:
- јака пигментација коже на јагодицама, пазуху и гениталијама;
- мрље одеће када је у контакту са знојним деловима тела;
- напади опште слабости;
- промуклим гласом.
Треба имати на уму да су алкаптонурија и охроноза, као што је горе наведено, синоними за исти поремећај катаболизма тирозина.
Болест јаворовог сирупа у урину и алкаптонурија. Конгенитална болест јаворовог сирупа у урину или леуциноза је такође метаболички поремећај, има исти образац наслеђивања, па чак и мутације се јављају на истом хромозому, али утичу на ген који кодира ензимски комплекс разгранатог ланца α-кето киселине дехидрогеназе. Због тога тело не може да разгради одређене компоненте протеина, посебно аминокиселине леуцин, изолеуцин и валин. Код ове болести, урин (и ушни восак) имају сладак мирис; поред тога, клиничка слика ове врсте органске ацидемије укључује хипопигментацију, флуктуације крвног притиска, нападе, повраћање и дијареју, пад нивоа глукозе у крви, кетоацидозу, халуцинације итд. Стопа смртности код деце је прилично висока; код одраслих, без лечења, може доћи до коме и смрти услед церебралног едема.
Албинизам и алкаптонурија су „уједињени“ само тирозином. Албинизам, укључујући окулокутани, узрокован је генетским мутацијама које утичу на производњу пигмента меланина. Конгениталне промене су примећене у гену TYR на хромозому 11 (11q14.3), који кодира тирозиназу, меланозомен ензим који садржи бакар, неопходан за формирање пигмента коже на основу продуката метаболизма тирозина. Ова болест је много чешћа од алкаптонурије.
Компликације и посљедице
Узрокована деловањем интермедијарних метаболита тирозина – хомогентизинске и бензохинон-сирћетне киселине – последице и компликације алкаптонурије настају услед таложења реактивних пигментираних полимера, уништавања колагенских фибрила и погоршања стања хрскавице (са смањењем њихове отпорности на механички стрес).
Током година, у одраслом добу, развијају се дегенеративни артритис и остеоартритис великих зглобова (кука, сакроилијачног и колена); интервертебрални простори се сужавају (посебно у лумбалној и торакалној кичми) – са калцификацијом и формирањем остеофита; густина ткива субхондралних коштаних плоча се смањује, а кости које се налазе испод њих могу проћи кроз патолошко ремоделирање са формирањем израслина и деформација. [ 11 ]
Може се приметити оштећење срчаних залистака (аортних и митралних) и коронарних артерија – са знацима коронарне болести срца, као и стварање камена у бубрезима и простати – због исте калцификације. [ 12 ], [ 13 ]
Дијагностика алкаптонурија
Типично, дијагноза конгениталних метаболичких поремећаја заснива се на проучавању биолошких течности тела.
На основу којих тестова и реакција се може дијагностиковати алкаптонурија? Потребни су тестови урина за откривање хомогентизинске киселине и одређивање њеног нивоа (нормално – 20-30 мг дневно, повишено – 3-8 г). Узорак урина се испитује гасном хроматографијом или масеном спектрометријом, користећи течну хроматографију; могућ је скрининг тест на присуство гвожђе хлорида у урину. [ 14 ]
Постоји и метода за брзу дијагностику – одређивање алкаптона у осушеним мрљама урина на папиру (по интензитету боје).
Приликом разјашњавања дијагнозе, инструментална дијагностика (радиографија) подразумева идентификацију радиолошких знакова остеоартритиса и других зглобних патологија код пацијената.
Дијагноза се потврђује молекуларно-генетским методама дијагностиковања наследних болести, као што су генетско тестирање и секвенцирање ДНК. [ 15 ]
Диференцијална дијагноза
Диференцијална дијагноза обухвата хемохроматозу и акутну инсуфицијенцију јетре новорођенчета, меланинурију, акутну интермитентну порфирију, хемофагоцитну лимфохистиоцитозу, примарну митохондријалну патологију, реуматоидни артритис, анкилозни спондилитис.
Кога треба контактирати?
Третман алкаптонурија
Главни третман за алкаптонурију је орална примена великих доза (најмање 1000 мг дневно) аскорбинске киселине. Код деце ово повећава излучивање хомогентизинске киселине урином, а код одраслих смањује садржај њеног деривата, бензохинонсирћетне киселине, у урину и успорава његово везивање за структуре везивног ткива зглобова и колагена. [ 16 ]
Западноевропске клинике тестирају лек Нитизинон (Орфалин), лек из групе метаболита који инхибира другу фазу катаболизма тирозина: трансформацију пара-хидроксифенилпирувата у хомогентизинску киселину. Међутим, употреба овог фармаколошког средства доводи до акумулације тирозина и може изазвати тешке нежељене ефекте, укључујући замућење рожњаче и фотофобију, крварење из носа и желуца, отказивање јетре, промене у крви итд. Ипак, у Сједињеним Државама, Нитизинон је одобрен од стране FDA за лечење тирозинемије типа I. [17 ], [ 18 ]
Стога се за проблеме са зглобовима узроковане алкаптонуријом спроводи физиотерапија – терапија вежбањем за повећање мишићне снаге и побољшање покретљивости зглобова, балнеотерапија и пелоидна терапија за ограничавање бола.
Иако се тирозин не уноси само храном већ се и производи у телу, пацијентима са алкаптонуријом се препоручује да се придржавају дијете са ниским садржајем протеина и да ограниче конзумацију хране богате тирозином, пре свега говедине и свињетине, млечних производа (посебно сирева), махунарки, орашастих плодова и семенки.
Превенција
Спречавање генских мутација је немогуће, али да би се спречило рађање деце са високим ризиком од урођених поремећаја, постоји медицинско генетско саветовање, које је неопходно пре планиране трудноће за оне парове чија породична историја укључује наследне болести. [ 19 ]
Прогноза
Смртни исходи алкаптонурије су веома ретки, а смрт може бити узрокована озбиљним компликацијама које укључују срце и бубреге. Дакле, укупни животни век људи са алкаптонуријом је добар.
Али квалитет живота је смањен због интензивног бола у зглобовима или кичми са значајним ограничењем покретљивости, често прогресивним.