
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Лејина субакутна некротизирајућа енцефаломиопатија
Медицински стручњак за чланак

Последње прегледано: 04.07.2025
 [
[ Узроци Лејиног синдрома
Болест се заснива на недостатку ензима који обезбеђују производњу енергије углавном због поремећаја метаболизма пирувичне киселине и дефекта у транспорту електрона у респираторном ланцу. Развија се недостатак комплекса пируват дехидрогеназе (а-Е1 подјединица), пируват карбоксилазе, комплекса 1 (НАД-коензим Q-редуктаза) и комплекса 4 (цитохром оксидаза) респираторног ланца.
Утврђено је да се дефекти пируват карбоксилазе, комплекса 1 (НАД-коензим Q-редуктаза) и комплекса 4 (цитохром оксидаза) респираторног ланца наслеђују аутозомно рецесивно, дефекти пируват дехидрогеназног комплекса (а-Е1 подјединица) наслеђују се Х-везано рецесивно. У случају тачкастих мутација мтДНК, које утичу на 6. подјединицу АТПазе, типично је митохондријално наслеђивање. Најчешће се јавља мисценс мутација, повезана са заменом тимина гванином или цитозином на позицији 8993 мтДНК. Ређе је мутација на позицији 9176 мтДНК. Због чињенице да је мутација T8993G главни дефект код НАРП синдрома, описане су породице са ове две болести. Код деце је описана и мутација у мтДНК на позицији 8344, која се јавља код МЕРФ синдрома.
Претпоставља се да се у случају акумулације мутантне мтДНК у већини митохондрија развија тежак ток Лијевог синдрома. У митохондријалној генези овог стања, мутантна мтДНК се налази у 90% свих митохондрија. Патогенеза је повезана са кршењем формирања енергије у ћелијама и развојем лактатне ацидозе.
Симптоми Лејиног синдрома
Први знаци болести се јављају у раном узрасту (1-3 године). Међутим, познати су случајеви манифестације болести већ у 2 недеље и у 6-7 година. У почетку се развијају неспецифични поремећаји: успорени психомоторни развој, смањен апетит, епизоде повраћања, дефицит телесне тежине. Накнадно се повећавају неуролошки симптоми: мишићна хипотонија или дистонија са преласком у хипертонију, напади миоклонуса или тонично-клоничних нападаја, тремор екстремитета, хореоатетоза, поремећај координације, смањени тетивни рефлекси, летаргија, поспаност. Церебрална неуродегенерација је прогресивна. Појачавају се симптоми пирамидалне и екстрапирамидалне инсуфицијенције, оштећен је чин гутања. Често се примећују промене у органу вида као што су птоза, офталмоплегија, атрофија оптичких живаца, ређе пигментна дегенерација мрежњаче. Понекад се развија хипертрофична кардиомиопатија, јављају се епизоде тахипнеје.
Ретко, болест се одвија као акутна енцефалопатија. Типичнији је хронични или субакутни ток, који доводи до смртног исхода неколико година након почетка болести. Код брзог тока (неколико недеља), смрт наступа као последица парализе респираторног центра.
Дијагностика Лејиног синдрома
Биохемијски тест крви открива лактатну ацидозу услед акумулације млечне и пирувичне киселине у крви и цереброспиналној течности, као и повећање садржаја аланина у крви. Ниво кетонских тела такође може бити повишен. У урину се открива повећано излучивање органских киселина: млечне, фумарне итд. Ниво карнитина у крви и ткивима се често смањује.
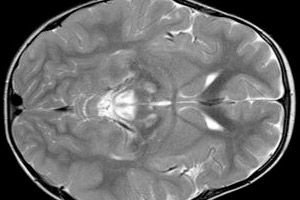
Резултати ЕЕГ-а откривају фокалне знаке епилептичке активности. Подаци МРИ откривају увећање можданих комора, билатерално оштећење мозга, калцификацију базалних ганглија (каудатно једро, путамен, субстанција нигра, глобус палидус). Такође се може открити атрофија можданих хемисфера и мождане масе.
Морфолошки преглед открива грубе промене у можданој материји: симетричне жаришта некрозе, демијелинације и сунђерасте дегенерације мозга, углавном средњих делова, моста, базалних ганглија, таламуса и оптичког нерва. Хистолошка слика обухвата цистичну дегенерацију можданог ткива, астроцитну глиозу, смрт неурона и повећање броја митохондрија у ћелијама. У скелетним мишићима долази до акумулације липидних инклузија, смањења хистохемијске реакције на комплексе 1 и 4 респираторног ланца, субсарколемалне акумулације митохондрија, абнормалних митохондрија са дезорганизацијом криста. Феномен РРФ се често не детектује.
Како испитивати?
Који су тестови потребни?