
Сви иЛиве садржаји су медицински прегледани или проверени како би се осигурала што већа тачност.
Имамо стриктне смјернице за набавку и само линкамо на угледне медијске странице, академске истраживачке институције и, кад год је то могуће, медицински прегледане студије. Имајте на уму да су бројеви у заградама ([1], [2], итд.) Везе које се могу кликнути на ове студије.
Ако сматрате да је било који од наших садржаја нетачан, застарио или на неки други начин упитан, одаберите га и притисните Цтрл + Ентер.
Ушеров синдром
Медицински стручњак за чланак

Последње прегледано: 04.07.2025
Ушеров синдром је наследна болест која се манифестује као потпуна глувоћа од рођења, као и прогресивно слепило са годинама. Губитак вида је повезан са ретинитис пигментозом, процесом пигментне дегенерације мрежњаче. Многи људи са Ушеровим синдромом такође имају тешке проблеме са равнотежом.
Епидемиологија
Захваљујући истраживању, било је могуће утврдити да Ушеров синдром погађа око 8% испитане глувонеме деце (тестови су спроведени у посебним установама за глувонеме особе). Пигментни ретинитис је примећен код 6-10% пацијената који пате од конгениталне глувоће, што се, заузврат, примећује код око 30% људи са пигментном болешћу мрежњаче.
Верује се да се ова болест манифестује код приближно 3-10 људи од 100 хиљада широм света. Може се подједнако јавити и код жена и код мушкараца. Око 5-6% светске популације пати од овог синдрома. Око 10% свих случајева тешке глувоће код деце јавља се због Ушеровог синдрома I, као и II типа.
У Сједињеним Државама, типови 1 и 2 су најчешћи типови. Заједно, они чине приближно 90 до 95 процената свих случајева Ушеровог синдрома код деце.
Узроци Ушеров синдром
Ушеров синдром типова I, II и III има аутозомно рецесивни узрок, док се тип IV сматра поремећајем X-хромозома. Узроци слепила и глувоће који се јављају код овог синдрома још увек нису довољно проучени. Претпоставља се да су људи са овом болешћу преосетљиви на компоненте које могу оштетити структуру ДНК. Поред тога, ова болест може бити повезана са поремећајима имуног система, али у овом случају не постоји тачна слика овог процеса.
Године 1989, хромозомске абнормалности су први пут идентификоване код пацијената са болешћу типа II, што би у будућности могло довести до начина изоловања гена који узрокују синдром. Такође би могло бити могуће идентификовати ове гене код носилаца и развити посебне пренаталне генетске тестове.
 [ 8 ]
[ 8 ]
Фактори ризика
Синдром се наслеђује када су оба родитеља погођена, тј. наслеђује се рецесивним типом. Дете такође може наследити болест ако су му родитељи носиоци гена. Ако оба будућа родитеља имају овај ген, онда је вероватноћа да ће имати бебу са овим синдромом 1 према 4. Особа која има само један ген за синдром сматра се носиоцем, али нема симптоме поремећаја. Данас још увек није могуће утврдити да ли особа има ген за ову болест.
Ако се дете роди од родитеља, од којих један нема такав ген, онда је вероватноћа да ће наследити синдром веома мала, али ће дефинитивно бити носилац.
Симптоми Ушеров синдром
Симптоми Ушеровог синдрома укључују губитак слуха и абнормално накупљање пигментираних ћелија у структурама ока. Пацијент затим развија дегенерацију мрежњаче, што узрокује погоршање вида и на крају губитак вида у најтежим случајевима.
Сензоринеурални губитак слуха може бити благ или потпун и обично не напредује од рођења. Међутим, болест пигментације мрежњаче може почети да се развија у детињству или касније. Резултати тестова су показали да се централна оштрина вида може одржати дуги низ година, чак и када се периферни вид погорша (стање које се назива „тунелски вид“).
То су главне манифестације болести, које понекад могу бити допуњене другим поремећајима, као што су психоза и други ментални поремећаји, проблеми са унутрашњим ухом и/или катаракта.
Обрасци
Током истраживања идентификоване су 3 врсте ове болести, као и 4. облик, који је прилично редак.
Тип I болести карактерише се урођеном потпуном глувоћом, као и поремећајем равнотеже. Често таква деца почињу да ходају тек са 1,5 година. Погоршање вида обично почиње са 10 година, а коначни развој стања ноћног слепила почиње са 20 година. Деца са овом врстом болести могу развити прогресивно погоршање периферног вида.
Код болести типа II, примећује се умерена или конгенитална глувоћа. У овом случају, погоршање делимичне глувоће често се више не јавља. Пигментни ретинитис почиње да се развија око краја адолесценције или након 20 година. Развој ноћног слепила обично почиње између 29 и 31 године. Оштећење оштрине вида код патологије типа II генерално напредује мало спорије него код типа I.
Тип III болести карактерише прогресивни губитак слуха, обично почевши током пубертета, као и постепени развој у истом периоду (нешто касније од губитка слуха) пигментозе ретинитиса, који може постати фактор у развоју прогресивног слепила.
Манифестације патологије типа IV углавном се јављају код мушкараца. У овом случају се такође примећују прогресивни поремећаји и губитак слуха и вида. Овај облик је веома редак и обично има X-хромозомску природу.
Дијагностика Ушеров синдром
Дијагноза Ушеровог синдрома се поставља на основу пацијентове примећене комбинације изненадне глувоће и прогресивног губитка вида.
Тестови
Може се наручити посебан генетски тест за откривање мутације.
Пронађено је једанаест генетских локуса који могу изазвати развој Ушеровог синдрома, а идентификовано је девет гена који су дефинитивно узрок поремећаја:
- Тип 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Тип 2: ush2a, VLGR1, WHRN.
- Ушеров синдром тип 3: USH3A.
Научници NIDCD-а, заједно са колегама са универзитета у Њујорку и Израелу, идентификовали су мутацију названу R245X у гену Pcdh15 која чини велики проценат Ушеровог синдрома типа 1 у јеврејској популацији.
Да бисте сазнали више о лабораторијама које спроводе клиничка испитивања, посетите https://www.genetests.org и потражите у директоријуму лабораторија термин „Ушеров синдром“.
Да бисте сазнали више о постојећим клиничким испитивањима која укључују генетско тестирање за Ушеров синдром, посетите https://www.clinicaltrials.gov и потражите „Ушеров синдром“ или „генетско тестирање Ушеровог синдрома“.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Инструментална дијагностика
Постоји неколико метода инструменталне дијагностике:
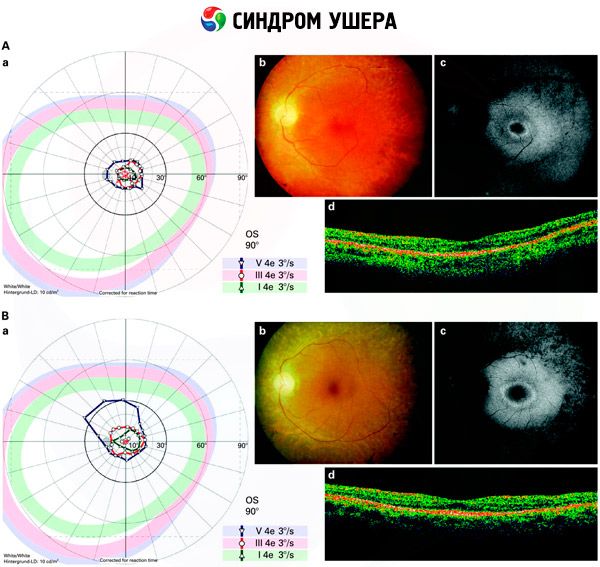
- Преглед фундуса ради откривања присуства пигментних мрља на мрежњачи, као и сужавања крвних судова мрежњаче;
- Електроретинограм, који омогућава откривање почетних дегенеративних одступања у мрежњачи ока. Показује изумирање електрорадиографских путева;
- Електронистагмограм (ЕНГ) мери невољне покрете очију који би могли указивати на присуство неравнотеже.
- Аудиометрија, која се користи за утврђивање присуства глувоће и њеног степена тежине.
Диференцијална дијагноза
Ушеров синдром мора се разликовати од неких сличних поремећаја.
Халгренов синдром, који карактерише конгенитални губитак слуха и прогресивни губитак вида (развијају се и катаракта и нистагмус). Додатни симптоми укључују атаксију, психомоторне поремећаје, психозу и менталну ретардацију.
Алстромов синдром, који је наследна болест код које ретина дегенерише, што доводи до губитка централног вида. Овај синдром је повезан са гојазношћу у детињству. Истовремено, дијабетес мелитус и губитак слуха почињу да се развијају након 10 година.
Рубеола код труднице у првом тромесечју може изазвати разне абнормалности у развоју детета. Међу последицама такве абнормалности су губитак слуха, као и (или) проблеми са видом, а поред овога и разни развојни дефекти.
Кога треба контактирати?
Третман Ушеров синдром
Тренутно не постоји лек за Ушеров синдром. Стога се терапија у овом случају углавном састоји од успоравања процеса губитка вида, као и од надокнађивања губитка слуха. Могуће методе лечења укључују:
- Узимање витамина А (неки офталмолози верују да високе дозе палмитата витамина А могу успорити, али не и зауставити, прогресију пигментозе ретинитиса);
- Имплантација посебних електронских уређаја у уши пацијента (слушни апарати, кохлеарни имплантати).
Офталмолози препоручују да већина одраслих са уобичајеним облицима пигментозе ретинитиса узима 15.000 ИЈ (интернационалних јединица) витамина А палмитата дневно под надзором. Пошто особе са Ушеровим синдромом типа 1 нису биле укључене у студију, високе дозе витамина А се не препоручују за ову групу пацијената. Људи који размишљају о узимању витамина А требало би да разговарају о овој опцији лечења са својим лекаром. Остале препоруке за ову опцију лечења укључују:
- Промените исхрану тако да укључите храну богату витамином А.
- Жене које планирају трудноћу треба да престану да узимају високе дозе витамина А три месеца пре планираног зачећа због повећаног ризика од урођених мана.
- Жене које су трудне треба да престану да узимају високе дозе витамина А због повећаног ризика од урођених мана.
Такође је важно прилагодити такво дете друштвеном животу. За то је потребна помоћ наставника специјалног образовања и психолога. У случају да је пацијент почео да доживљава прогресиван губитак вида, треба га научити да користи знаковни језик.
Прогноза
Ушеров синдром има неповољну прогнозу. Видно поље и његова оштрина почињу да се погоршавају у периоду од 20-30 година код већине пацијената са овом болешћу било које врсте. У неким случајевима долази до потпуног билатералног губитка вида. Губитак слуха, који је увек праћен немом, веома брзо се развија до потпуног билатералног губитка слуха.